第四章_芳环上的取代反应
- 格式:ppt
- 大小:1.43 MB
- 文档页数:74
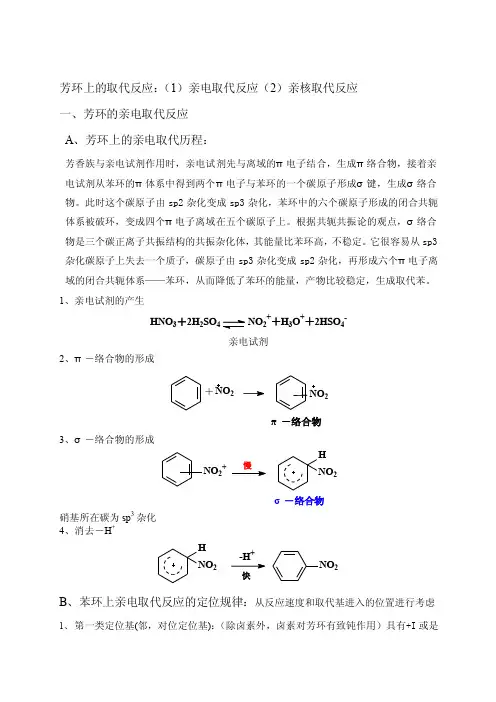
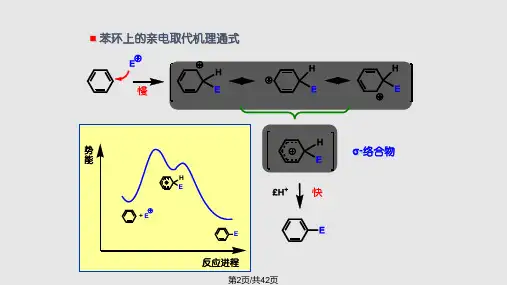
芳环上的取代反应:(1)亲电取代反应(2)亲核取代反应 一、芳环的亲电取代反应 A 、芳环上的亲电取代历程:芳香族与亲电试剂作用时,亲电试剂先与离域的π电子结合,生成π络合物,接着亲电试剂从苯环的π体系中得到两个π电子与苯环的一个碳原子形成σ键,生成σ络合物。
此时这个碳原子由sp2杂化变成sp3杂化,苯环中的六个碳原子形成的闭合共轭体系被破环,变成四个π电子离域在五个碳原子上。
根据共轭共振论的观点,σ络合物是三个碳正离子共振结构的共振杂化体,其能量比苯环高,不稳定。
它很容易从sp3杂化碳原子上失去一个质子,碳原子由sp3杂化变成sp2杂化,再形成六个π电子离域的闭合共轭体系——苯环,从而降低了苯环的能量,产物比较稳定,生成取代苯。
1、亲电试剂的产生HNO 3+2H 2SO4NO 2++H 3O ++2HSO 4-亲电试剂2、π-络合物的形成+NO 2π-络合物23、σ-络合物的形成NO 2+HNO2σ-络合物硝基所在碳为sp 3杂化 4、消去-H ++NO 2H NO 2快B 、苯环上亲电取代反应的定位规律:从反应速度和取代基进入的位置进行考虑1、 第一类定位基(邻,对位定位基):(除卤素外,卤素对芳环有致钝作用)具有+I 或是+C 效应,其作用是增大芳环的电子云密度。
致活基NH 2NHR2OHORNHCROPhR致钝基F Cl BrI2、 第二类定位基(间位定位基):具有-I 或-C 效应,使芳环上的电子云密度降低,均为致钝基NO 2NR 3COOHCOORSO 3HCNCHOCROCCl 3C 、影响亲电取代的因素:(1)芳环上取代基对于E +进入芳环位置的影响第一类定位基-邻对位定位基第二类定位基-间位定位基共振式越多, 正电荷分散程度越大,芳正离子越稳定。
(2) 动力学控制与热力学控制: α位取代-动力学控制产物; β位取代-热力学控制产物。
(3) 邻位和对位定向比:a 亲电试剂的活性越高,选择性越低。






![芳环上的磺化取代[终稿]](https://uimg.taocdn.com/77a7a92d590216fc700abb68a98271fe910eaf6f.webp)
芳环上的取代磺化一、磺化反应的历程及动力学磺化反应:一种向有机分子中引入磺酸基(—SO3H)或磺酰氯基(—SO3Cl)的反应过程,磺化是磺基(或磺酰卤基)中的硫原子与有机分子中的碳原子相连接形成C-S键的反应,生成的产物是磺酸(R—SO3H,R表示烃基)、磺酸盐(R—SO3M;M表示NH4或金属离子)或磺酰氯(R—SO2Cl)。
在这儿着重介绍芳环上的取代磺化。
磺化是亲电取代反应,因此芳环上有供电基使磺化反应速率变快,有吸电基使磺化反应速率变慢。
磺化也是连串反应,但是与氯化不同,磺酸基对芳环有较强的钝化作用,一磺酸比相应的被磺化物难于磺化,而二磺酸又比相应的一磺酸难于磺化。
因此,苯系和萘系化合物在磺化时,只要选择合适的反应条件,例如磺化剂的浓度和用量、反应的温度和时间,在一磺化时可以使被磺化物基本上完全一磺化,只副产很少量的二磺酸;在二磺化时只副产很少量的三磺酸。
例如,在苯的共沸去水一磺化时,磺化液中约含有88%~91%苯磺酸、小于 15%苯、小于0.5%苯二磺酸和2.0%~4%硫酸(均为质量分数,下同)。
芳香化合物进行磺化反应时,分两步进行。
首先,亲电质点向芳环进行亲电进攻,生成σ-配合物,后在碱作用下脱去质子得到芳环酸。
反应历程如下:研究证明,用浓硫酸磺化时,脱质子较慢,第二步是整个反应速率的控制步骤。
用稀酸磺化时,生成σ-配合物较慢,第一步限制了整个反应速率。
采用发烟硫酸或硫酸磺化芳烃时,其反应动力学可如下表示。
当磺化质点为SO3 时:v=kso3[ArH][SO3]=k′SO3[ArH][H2O]2-当磺化质点为H2S2O7时:v=kSA[ArH][H2S2O7]=k′H2S2O7[ArH][H2O]2-当磺化质点为H3SO4+时:v=kH3SO4 +[ArH][H3SO4+]=k′H3SO4 +[ArH][H2O]-由以上三式可以看出,磺化反应速率与磺化剂中的含水量有关。
当以浓硫酸为磺化剂,水很少时,磺化反应速率与水浓度的平方成反比,即生成的水量越多,反应速率下降越快。

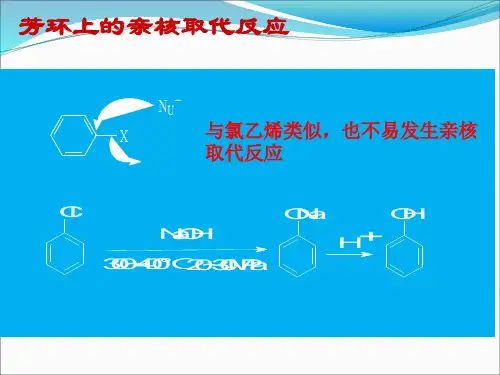
芳环上的取代氯化反应
芳环取代氯化反应是一种常见的有机化学反应,用于在芳环分子
的位置上引入氯原子。
该反应通常以氯化剂和芳环化合物为反应物,经过催化剂的作用,氯元素被取代到芳环化合物的碳原子上。
在常见的芳环取代氯化反应中,常用的氯化剂包括氯化亚铁(III)、氯化铁(III)、氯化亚锡(IV)等,催化剂常见的有氯化铜(II)、三氯化铝、五氯化钒等。
反应过程中,氯化剂首先被还原为氯离子,然后与芳环化合物发
生取代反应。
反应中,催化剂起到催化作用,降低了反应的活化能。
催化剂通常通过与氯离子形成络合物,加快了反应速率。
反
应的基本机理是亲电取代机理,即氯离子通过亲电攻击芳环化合物,将氯原子引入到芳环位置上。
芳环取代氯化反应的反应条件可以根据具体的反应物来确定。
通常,在适当的溶剂中进行反应,并控制反应温度和反应时间,以
获得较好的产率和选择性。
通过芳环取代氯化反应,可以在芳环化合物的分子结构上引入氯
原子,改变其物化性质以及反应性质。
此外,芳环取代氯化反应
还可用于合成具有特定结构的有机化合物,如取代基含有氯原子
的药物、杀虫剂等。
芳环取代氯化反应是一种常用的有机化学反应,能够在芳环化合
物的位置上引入氯原子。
该反应需要适当的氯化剂和催化剂,同
时在合适的条件下进行,以获得较好的反应产率和选择性。