B2 无菌制剂生产管理
- 格式:ppt
- 大小:4.27 MB
- 文档页数:111

附录1:无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括注射剂、眼用制剂、无菌软膏剂、无菌混悬剂等。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第三条悬浮粒子、浮游菌、沉降菌和表面微生物等测试方法应按照相关标准执行。
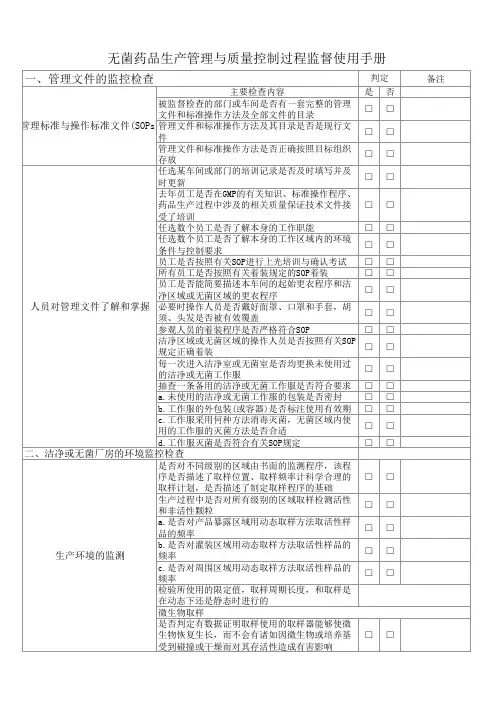
第二章原则第四条无菌药品的生产须满足其质量和预定用途的要求,应最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验。
第五条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第六条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,如采用机械连续传输物料时,应采用正压气流保护并监测压差。
物料准备、产品配制和灌装或分装等操作必须在洁净区内分区(室)进行。
第七条应按所需环境的特点确定无菌药品洁净生产区的级别。
每一步生产操作的环境都应达到适当的动态洁净度标准,以尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下 4 个级别:A 级高风险操作区,如:灌装区、放置胶塞桶、敞口安瓿瓶、敞口西林瓶的区域及无菌装配或连接操作的区域。
通常用单向流操作台(罩)来维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应有数据证明单向流的状态并须验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级指无菌配制和灌装等高风险操作 A 级区所处的背景区域。
C 级和D 级指生产无菌药品过程中重要程度较低的洁净操作区。
以上各级别空气悬浮粒子的标准规定如下1表:注:(1) 为了确定A级区的级别,每个采样点的采样量不得少于1m3。

无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
注:(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。

附录1:无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
以上各级别空气悬浮粒子的标准规定如下表:注:(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。

![《药品生产质量管理规范》(2010年修订)附录[参考]](https://uimg.taocdn.com/1eb0010b80eb6294dc886c7f.webp)
关于发布《药品出产质量办理规范(2010年修订)》无菌药品等5个附录的布告2011年02月24日发布国家食品药品监督办理局公告2011年第16号关于发布《药品出产质量办理规范(2010年修订)》无菌药品等5个附录的布告有关办理事宜的布告根据卫生部令第79号《药品出产质量办理规范(2010年修订)》第三百一十条规则,现发布无菌药品、质料药、生物制品、血液制品及中药制剂等5个附录,作为《药品出产质量办理规范(2010年修订)》配套文件,自2011年3月1日起实施。
特此布告。
附件:1.无菌药品2.质料药3.生物制品4.血液制品5.中药制剂国家食品药品监督办理局二○一一年二月二十四日附录1:无菌药品第一章规模第一条无菌药品是指法定药品规范中列有无菌查看项意图制剂和质料药,包含无菌制剂和无菌质料药。
第二条本附录适用于无菌制剂出产全进程以及无菌质料药的灭菌和无菌出产进程。
第二章原则第三条无菌药品的出产须满意其质量和预订用处的要求,应当最大极限下降微生物、各种微粒和热原的污染。
出产人员的技能、所承受的训练及其作业态度是到达上述方针的要害要素,无菌药品的出产有必要严厉依照精心规划并经验证的办法及规程进行,产品的无菌或其它质量特性绝不能只依托于任何办法的终究处理或制品查验(包含无菌查看)。
第四条无菌药品按出产工艺可分为两类:选用终究灭菌工艺的为终究灭菌产品;部分或悉数工序选用无菌出产工艺的为非终究灭菌产品。
第五条无菌药品出产的人员、设备和物料应经过气锁间进入洁净区,选用机械接连传输物料的,应当用正压气流保护并监测压差。
第六条物料预备、产品制造和灌装或分装等操作有必要在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等要素,承认无菌药品出产用洁净区的等级。
每一步出产操作的环境都应当到达恰当的动态洁净度规范,尽或许下降产品或地址理的物料被微粒或微生物污染的危险。
第三章洁净度等级及监测第八条洁净区的规划有必要契合相应的洁净度要求,包含到达“静态”和“动态”的规范。

无菌制剂生产质量管理规范2004年9月 美国FDA发布2009年6月 药审中心组织翻译 武田药品有限公司翻译北核协会审核药审中心最终核准目录Ⅰ. 前言 (1)Ⅱ. 背景 (1)A.规章制度 (1)B.技术框架 (2)Ⅲ. 范围 (2)Ⅳ. 厂房和设施 (3)A.关键区域-100级(ISO 5) (4)B.支持洁净区 (6)C.洁净区隔离 (6)D.空气过滤 (7)1. 膜过滤 (7)2. 高效空气微粒过滤(HEPA) (7)E. 设计 (8)Ⅴ. 人员培训、资格和监督 (10)A.人员 (11)B.实验室人员 (13)C.监测计划 (13)Ⅵ. 组分和容器/密闭系统 (14)A.组分 (14)B.容器/密闭系统 (15)1. 准备 (15)2. 容器密封系统的检测 (16)Ⅶ. 内毒素控制 (17)Ⅷ. 时间限度 (18)Ⅸ. 无菌操作和灭菌的验证 (18)A. 操作过程模拟 (19)1. 研究设计 (19)2. 运行频率和次数 (20)3. 运行时间 (20)4. 运行量 (21)5. 线速度 (21)6. 环境条件 (21)7. 培养基 (22)8. 灌装培养基装置的培养和检查 (22)9. 试验结果判断 (23)B. 过滤效率 (24)C. 设备、容器和密闭系统的灭菌 (26)1. 合格证明和验证 (26)2. 设备控制和仪器校正 (27)Ⅹ. 实验室控制 (28)A. 环境监测 (29)1. 一般书面计划 (29)2. 确定水平和倾向计划 (30)3. 消毒效果 (31)4. 监测方法 (31)B. 微生物培养基和鉴定 (32)C. 粗滤生物负荷 (33)D. 替代微生物学检测方法 (33)E. 微粒监测 (33)Ⅺ. 无菌检测 (33)A. 微生物学实验室控制 (35)B. 取样和孵育 (35)C. 无菌阳性的调查 (35)Ⅻ. 批记录审核:工艺控制文件 (37)附件1:无菌操作隔离室.......................................................................................1~4 附件2:吹-灌装-密封技术................................................................................... 1~2 附件3:灌装和密封操作前的处理........................................................................ 1~2 参考文献相关指导原则文件术语汇编无菌制剂生产质量管理规范Ⅰ.前言本指导原则旨在帮助制造商在用无菌操作生产无菌药物和生物制品时符合管理机构现行药品生产质量管理规范(CGMP)的规定(2l CFR 210 和 211部分)。

无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。
第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
注:(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。

药品无菌生产注意事项药品无菌生产是指在无菌条件下进行药品制造的过程。
无菌条件是指在产品接触的所有环境、设备、容器和材料均不存在或具备严格控制的微生物限度。
无菌生产的目的是确保药品的质量和安全性,防止微生物污染引起的感染。
在药品无菌生产过程中,需要严格遵循一系列注意事项,以确保生产过程的无菌性。
首先,必须建立合适的场所和设备。
无菌生产需要在无菌室或无菌区域进行,通常采用带正压或负压的洁净区域。
无菌室内的墙壁、地面和天花板应具备无菌性,并能够抵抗湿热、容易清洁和消毒。
设备应具备无菌性能,易于清洁和消毒,并能满足生产工艺的要求。
其次,必须建立适当的人员培训和操作规程。
所有参与无菌生产的人员必须经过培训,了解无菌生产的原理和操作规程。
人员需要佩戴合适的个人防护装备,如帽子、口罩、手套和无菌服,以防止微生物的外源性污染。
第三,必须进行严格的材料控制。
所有使用的原辅料和容器必须经过合格的供应商采购,并进行严格的检验和评估。
原辅料和容器必须满足适当的无菌性标准,并进行适当的处理和灭菌。
容器的灌装、封口和包装必须在无菌条件下进行,以确保产品的无菌性。
第四,必须实施严格的工艺控制。
药品无菌生产过程中的每个步骤都必须进行适当的控制和监测,以确保无菌条件的维持。
如灌装过程中,必须进行合适的消毒和灭菌操作,保证灌装设备的无菌性。
第五,必须实施适当的环境监测和控制。
在无菌生产过程中,必须对环境进行适当的监测,包括空气微生物数量、空气颗粒物和工作区表面的微生物数量。
必须定期进行验证,确保环境的无菌性符合要求。
第六,必须实施适当的清洁和消毒措施。
无菌生产过程中,必须对设备、容器和工作区域进行适当的清洁和消毒,以防止微生物交叉污染。
第七,必须进行适当的质量控制。
药品的成品必须经过严格的质量控制,包括产品的无菌性测试和微生物限度测试。
必须建立适当的质量控制标准和程序,确保产品的质量合格。
总之,药品无菌生产是一项复杂的过程,需要严格遵循一系列的注意事项。

药品生产质量管理规范(新版GMP)附录:无菌药品第一章范围第一条无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药第二条本附录适用于无菌制剂生产全过程以及无菌原料药的灭菌和无菌生产过程。
第二章原则第三条无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染。
生产人员的技能、所接受的培训及其工作态度是达到上述目标的关键因素,无菌药品的生产必须严格按照精心设计并经验证的方法及规程进行,产品的无菌或其它质量特性绝不能只依赖于任何形式的最终处理或成品检验(包括无菌检查)。
第四条无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
第五条无菌药品生产的人员、设备和物料应通过气锁间进入洁净区,采用机械连续传输物料的,应当用正压气流保护并监测压差。
第六条物料准备、产品配制和灌装或分装等操作必须在洁净区内分区域(室)进行。
第七条应当根据产品特性、工艺和设备等因素,确定无菌药品生产用洁净区的级别。
每一步生产操作的环境都应当达到适当的动态洁净度标准,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
第三章洁净度级别及监测第八条洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
第九条无菌药品生产所需的洁净区可分为以下4个级别:A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
以上各级别空气悬浮粒子的标准规定如下表:新版GMP附录无菌药品注:(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。
一、总则为确保医药公司生产、销售和使用过程中无菌物品的安全与质量,防止微生物污染,保障人民群众用药安全,特制定本制度。
二、无菌物品的定义无菌物品是指在特定条件下,经过物理或化学方法处理的物品,其表面、内部和包装材料均无活微生物存在。
三、无菌流程管理制度1. 无菌物品的采购与验收(1)采购部门应选择具备相应资质的无菌物品供应商,并签订质量保证协议。
(2)采购的无菌物品应具有合格证明文件,如产品合格证、检验报告等。
(3)验收部门对采购的无菌物品进行严格验收,确保物品符合规定要求。
2. 无菌物品的储存与保管(1)无菌物品应存放在清洁、干燥、通风、避光的库房内。
(2)库房内应设有温湿度控制设备,确保库房内温湿度符合要求。
(3)无菌物品应分类存放,避免交叉污染。
(4)定期检查库房内无菌物品的储存条件,发现问题及时处理。
3. 无菌物品的灭菌与包装(1)采用物理或化学方法对无菌物品进行灭菌处理,确保灭菌效果。
(2)灭菌后的物品应进行包装,包装材料应符合无菌要求。
(3)包装过程中,操作人员应穿戴无菌手套、口罩等防护用品。
4. 无菌物品的使用(1)使用无菌物品前,应检查包装的完整性,如有破损或污染,不得使用。
(2)使用无菌物品时,应遵守无菌操作规程,防止微生物污染。
(3)无菌物品开启后,应尽快使用,不得长时间暴露在空气中。
(4)使用后的无菌物品应按照规定进行处理,防止交叉污染。
5. 无菌物品的运输与分发(1)运输无菌物品的工具应保持清洁、干燥,避免污染。
(2)运输过程中,应采取有效措施,防止无菌物品受到损坏或污染。
(3)分发无菌物品时,应按照规定程序进行,确保物品安全送达。
6. 无菌流程的监控与评估(1)建立无菌流程监控体系,定期对无菌流程进行检查、评估。
(2)对发现的问题及时进行整改,确保无菌流程的持续改进。
四、奖惩措施1. 对严格遵守无菌流程,确保产品质量的部门和个人给予奖励。
2. 对违反无菌流程,造成产品质量问题的部门和个人进行处罚。