武汉大学分子模拟实验作业第八章分子轨道模拟
- 格式:docx
- 大小:3.27 MB
- 文档页数:17

化学反应机理的计算模拟方法化学反应机理的计算模拟方法在现代化学研究中起着至关重要的作用。
通过计算模拟方法,研究人员可以深入了解化学反应中物质的转化过程和反应机理,以及研究反应速率、平衡常数和能量变化等关键参数。
本文章将详细介绍三种常见的化学反应机理计算模拟方法,包括量子力学方法、分子力场方法和轨道理论方法。
1. 量子力学方法:量子力学方法是一种基于量子力学原理的计算模拟方法,可以用于研究小分子和分子间相互作用。
其中,最常用的方法是密度泛函理论(DFT),它通过求解电子的波函数来计算能量和分子性质。
DFT方法可以准确地预测反应的能垒、中间体、过渡态和反应物与产物的力学和热力学性质。
此外,还有耦合簇方法(CC)、多配置自洽场(MCSCF)和多体微扰理论(MP2)等方法,它们可以考虑电子相关性,提高计算精度。
2. 分子力场方法:分子力场方法是一种基于力场原理的计算模拟方法,可以用于研究大分子和生物分子的结构和性质。
分子力场方法主要通过建立分子的力场参数来模拟分子的振动、转动和相互作用等过程。
其中,最常用的方法是力常数法、分子力常数法和AMBER力场等。
这些方法可以较好地预测大分子的构象、能量和动力学性质,并且计算速度较快,适合处理大尺寸体系。
3. 轨道理论方法:轨道理论方法是一种基于轨道与电子结构之间相互关系的计算模拟方法,可以研究分子的电子结构和反应机理。
其中,最常用的方法是分子轨道理论(MO)和密度泛函轨道理论(DFT)。
这些方法可以计算分子的轨道能级、轨道异构体的相对稳定性,以及反应物、产物和过渡态的电子结构参数。
轨道理论方法对于研究化学反应的电子过程和反应机理有重要意义。
除了以上三种常见的化学反应机理计算模拟方法,还有一些其他方法,比如过渡态搜索算法、动力学模拟方法和哈密顿路径法等,它们在特定的研究领域具有重要的应用价值。
在实际应用中,研究人员通常会根据具体问题选择合适的计算模拟方法,以获得准确的结果。

乙醛氧化生成乙酸反应机理的分子模拟夏垒;龙军;武志强;赵毅;代振宇;王立华【摘要】采用基于密度泛函理论(DFT)的量子化学方法研究了乙醛氧化生成乙酸的反应过程.结果表明:无氧条件下链引发过程最难发生,反应能垒达到380.78kJ/mol,但氧气可将此过程的反应能垒降至116.26kJ/mol;乙酰基自由基生成过氧乙酸以及链终止反应的各步骤的反应能垒均较低,反应较快;过氧乙酸转化为乙酸的反应较难发生,为整体反应过程的速率控制环节.此过程有2个可能的反应路径,速率控制步骤分别为过氧乙酸均裂生成乙酸自由基和羟基自由基的过程及过氧乙酸与乙醛反应生成乙醛单过氧乙酸酯中间化合物的过程,反应能垒分别为147.18、137.21kJ/mol.%Density functional theory (DFT) simulation method was applied to study the oxidization process of acetaldehyde to aceticacid.Simulation results suggest that, chain initiation step should be the most difficult step under anaerobic condition with a reaction energy barrier 380.78kJ/mol.However, with the presence of oxygen, the reaction energy barrier could be decreased to 116.26kJ/mol.It has been found that reaction energy barriers in each step of acetyl free radicals reacting to peracetic acid are quite low, and also in chain termination step.It is also noticed that the reaction of peracetic acid to acetic acid is difficult to take place, and it should be the rate control step in the overall reaction process.There are two possible reaction paths in the process.The reaction rate control steps could be homolytic cleavage of peracetic acid to generate acetic acid free radical and hydroxyl free radical, or the reaction of acetaldehyde with peracetic acid to produce acetaldehydemonooxyacetate.The reaction energy barriers of the above two reactions are 147.18kJ/mol and 137.21kJ/mol, respectively.【期刊名称】《石油学报(石油加工)》【年(卷),期】2019(035)001【总页数】8页(P20-27)【关键词】分子模拟;氧化;醛;羧酸【作者】夏垒;龙军;武志强;赵毅;代振宇;王立华【作者单位】中国石化石油化工科学研究院, 北京 100083;中国石化石油化工科学研究院, 北京 100083;中国石化石油化工科学研究院, 北京 100083;中国石化石油化工科学研究院, 北京 100083;中国石化石油化工科学研究院, 北京 100083;中国石化石油化工科学研究院, 北京 100083【正文语种】中文【中图分类】TE626醛类化合物是一类重要的有机原料及化学中间体,其可以通过还原反应制备醇类化合物[1-2],还可以通过氧化作用制备羧酸类化合物[3-5]。

竭诚为您提供优质文档/双击可除分子模型实验报告篇一:结构化学实验报告重庆大学化学化工学院《结构化学》实验报告姓名学号:年级专业:指导老师:重庆大学化学化工学院20XX年12月21日实验一利用量子化学计算软件验证分子轨道理论和判断分子点群一、主要仪器设备及软件1、仪器:用于计算的计算机。
2、软件:gviewA、建模软件(1)chemoffice是一款广受化学学习、研究者好评的化学学习工具。
(2)gaussView主要功能有创建三维分子模型,计算任务设置全面支持gaussian计算,和显示gaussian计算结果等。
b、计算软件:(1)gaussian:量子化学领域最著名和应用最广泛的软件之一,由量子化学家约翰波普的实验室开发,可以应用从头计算方法、半经验计算方法等进行分子能量和结构;过渡态能量和结构;化学键及反应能量;分子轨道;偶极矩;多极矩;红外光谱和拉曼光谱,核磁共振,极化率和超极化率,热力学性质,反应路径等分子相关计算。
(2)materialsstudio:是AcceLRYs公司专门为材料科学领域研究者所涉及的一款可运行在pc上的模拟软件。
(3)VAsp是使用赝势和平面波基组,进行第一定律分子动力学计算的软件包。
(4)gamess-us:由于免费与开放源码,成为除gaussian 以外,最广泛应用的量子化学软件,目前由Iowastateuinversity的markgorden教授的研究组主理。
(5)cAsTep:是由密度泛函理论为基础的计算程式所组成,同时采用平面波(planewave)为基底处理波函数,可针对具有周期性的固态材料表面进行化学模拟计算。
(6)ATK:是由丹麦公司QuantumwiseA/s开发的一款通用的电子态结构计算软件。
其他量子化学计算软件目前,除了上面提到的几版著名量子化学计算软件之外,还有大量商业和免费的量子化学计算软件,其中绝大部分是从事量子化学或计算化学研究的实验室自行开发的,此外,一些著名的大型化学软件如hyperchem、chem3D、sybyl等,也包含有量子化学计算包。
分子模拟实验报告
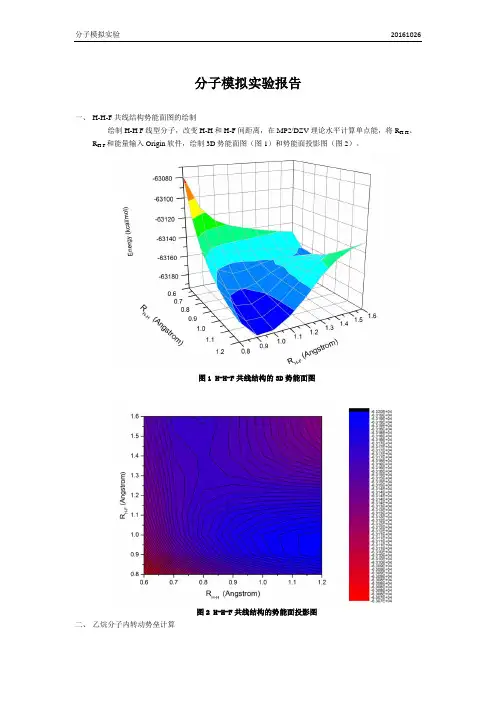
一、H-H-F共线结构势能面图的绘制
绘制H-H F线型分子,改变H-H和H-F间距离,在MP2/DZV理论水平计算单点能,将R H-H、R H-F 和能量输入Origin软件,绘制3D势能面图(图1)和势能面投影图(图2)。
图1 H-H-F共线结构的3D势能面图
图2 H-H-F共线结构的势能面投影图
二、乙烷分子内转动势垒计算
在Chem3d中画出NO分子,点击Surfaces-ChooseSurface-MolecularOrbital绘制分子轨道等值面图,通过SelectMolecularOrbital选择不同的分子轨道,截图如下。
LUMO+1 (163.115 eV)LUMO(-2.832 eV)
HOMO(-2.832 eV)HOMO-1 (-12.846 eV)
HOMO-2 (-21.326 eV)HOMO-3 (-21.326 eV)
HOMO-4 (-23.922 eV)HOMO-5 (-52.100 eV)
图3NO分子的八个分子轨道等值面图
按轨道的能量从低到高的顺序排列,使用Origin软件绘制NO的分子轨道能级图(图4)。
图3NO分子的分子轨道能级图
三、自我测评
本次实验继续练习了Chem3D中的扫描操作,学习了Origin软件中三维势能面图和势能面图的绘制和
Chem3D中分子轨道相关功能。
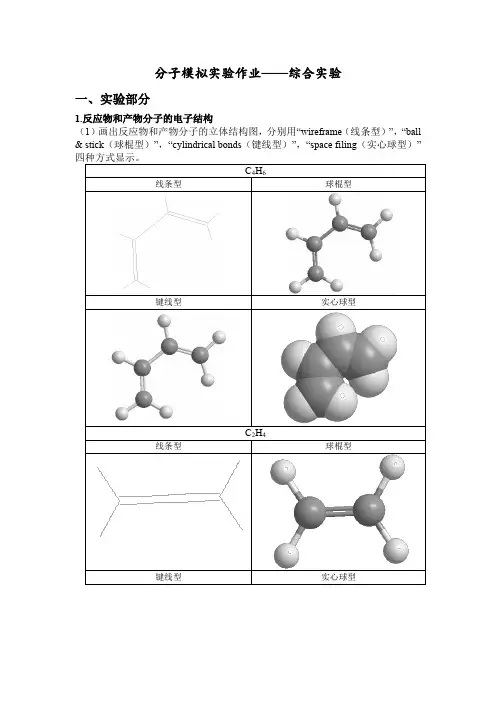
分子模拟实验作业——综合实验一、实验部分1.反应物和产物分子的电子结构(1)画出反应物和产物分子的立体结构图,分别用“wireframe(线条型)”,“ball & stick(球棍型)”,“cylindrical bonds(键线型)”,“space filing(实心球型)”(3)计算分子轨道,并图示各个分子的HOMO和LUMO轨道的形状和能量。
将所有的分子轨道按能级排列次序,并以此分析两反应物的轨道匹配情况。
C4H6LUMO HOMOC2H4LUMO HOMOC6H10LUMO HOMO反应是由1,3-丁二烯的HOMO与乙烯的LUMO反应,故图为分子轨道很匹配,有利于加成反应。
DA反应的反应物分为两部分,双烯体提供共轭双烯,亲双烯体提供不饱和键。
DA反应是由双烯体的HOMO与亲双烯体的LUMO发生作用,反应过程中,电子从双烯体的HOMO流入亲双烯体的LUMO。
C2H4的LUMO轨道与C4H6的HOMO轨道电子云相匹配,正好符合双烯合成的条件,利于反应的进行。
(4)绘制反应物和产物分子的总电子密度图和静电势图,分析两个反应物分子的电性匹配情况。
总电子密度图静电势图总电子密度图静电势图由轨道匹配来说,要使得反应能够顺利进行,必须是同为正或负。
C2H4的静电势图与C4H6的静电势,很匹配,所以有利于加成反应。
2.构象搜索与分子间长程相互作用(1)计算丁二烯分子绕CCCC二面角转动的构象,确定稳定构型,并计算内转动的能垒高度。
(可能不止一个)此时的构型有两种,顺式构型和反式构型。
稳定构型为反式构型,内转动能垒高度:40.58kcal/mol(反式构型)和38.67kcal/mol(顺式构型)。
图1 扫描势能面时的构型能量单位:kcal/mol①势能面图:图2丁二烯分子绕CCCC 二面角转动的构象②相互作用势能线及拟合图选取每个距离下的最低能量,减去MM2优化得到的C2H4和C4H6的能量 C2H4:Total: 0.4267 C4H6:Total: -0.1615 数据表格如下:CCCC 二面角/(°)质心距离/Å质心距离/Å相互作用能量(kc al/图3 C 4H 6与C 2H 4相互作用势能曲线3 反应途径计算(1)在HF/6-31G(d)理论水平上计算两个反应物分子和产物分子的298.15K时标准生成焓。
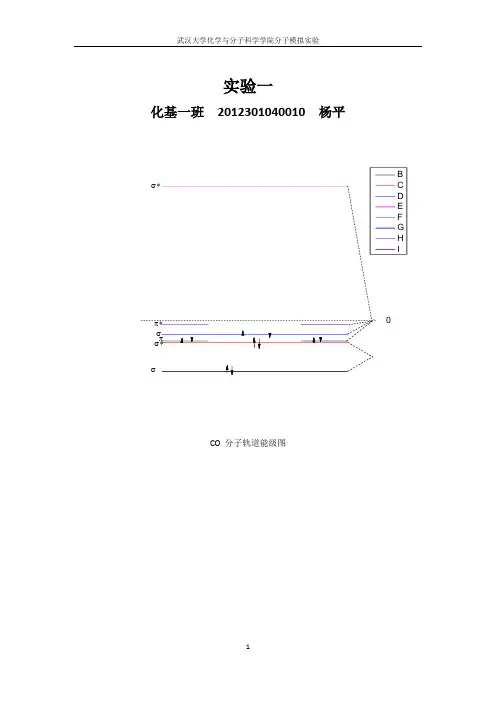
实验一
化基一班2012301040010 杨平
CO 分子轨道能级图
4
5
6
7
8
9
10
-70
-60-50-40-30-20-10010∆E /c m
-1
R/A
Lennard-Jones(12-6)势能函数拟合甲烷分子间势能曲线
感受与收获:
从第一次分子模拟实验中,了解到了分子模拟实验到底是一门什么样的课程。
学会运用计算机软件来帮助人们解决一些较为繁琐的数学等方面问题,以及运用软件来模拟化学反应过程,为实验室科学研究提供帮助。
第一次实验中主要是学习运用Origin 软件来帮助我们处理一些实验数据,通过正确的函数运用,成功模拟出了甲烷分子间势能曲线的方程。
还通过学习掌握了
一些基本的数据处理时候会用到的有用操作,都是一些实用性很强的操作,这些都为以后的数据处理带来了方便。
还有就是学了一些关于Chem3D 的一些基本操作,通过这款软件可以看出分子的一些物理常数,以及分子构型,这些都为以后的科学研究提供了帮助,很有学习意义。
分子模拟实验为我们更好的学习化学提供了另一片天地,我们很有必要好好去学习这一门课程。

引言概述:分子动力学模拟(MD)是一种模拟系统内原子或分子运动的计算方法,通过计算原子之间的相互作用力和运动方程,可以研究材料的物理和化学性质、相互作用和动态行为等。
本文将深入探讨分子动力学模拟的相关内容,包括模拟算法、分子模型构建、初始条件设定、系统参数调优、结果分析等。
正文内容:一、模拟算法1.1简单分子动力学模拟算法:介绍经典分子动力学模拟的基本原理和算法。
1.2高级模拟算法:介绍一些基于统计力学和量子力学原理的高级分子动力学模拟算法,如MonteCarlo方法和量子分子动力学模拟。
二、分子模型构建2.1原子选择:根据研究对象和目的,选择适合的原子种类。
2.2原子间相互作用模型:介绍常用的原子间相互作用势函数模型,如LennardJones势和Coulomb势等。
2.3拓扑构建:说明如何根据分子结构构建拓扑,包括原子连接方式和键长、键角、二面角等参数。
三、初始条件设定3.1初始构型:介绍如何原子或分子的初始位置和速度。
3.2温度控制:讨论如何在模拟中控制温度,包括使用温度计算公式和应用恒温算法等。
3.3压力控制:介绍如何在模拟中控制压力,包括应用压力计算公式和应用恒压算法等。
四、系统参数调优4.1时间步长选择:讲解如何选择合适的时间步长,以确保模拟结果的准确性和稳定性。
4.2模拟时间长度:介绍如何选取适当的模拟时间长度,以获得足够的统计样本。
4.3系统尺寸选择:探讨系统尺寸对模拟结果的影响,包括边界条件的选择和静电相互作用的处理。
五、结果分析5.1动力学参数计算:介绍如何通过模拟数据计算动力学参数,包括径向分布函数和速度自相关函数等。
5.2结构参数分析:讨论如何分析模拟结果中的结构特征,如配位数、键长分布和角度分布等。
5.3物理性质计算:讲解如何通过模拟数据计算材料的物理性质,如热力学性质和动力学性质等。
总结:分子动力学模拟是一种强大的计算工具,可以模拟和研究材料的动态行为和性质。
从模拟算法、分子模型构建、初始条件设定、系统参数调优到结果分析,每个步骤都需要仔细考虑和调整,以保证模拟结果的准确性和可靠性。

分子动力学模拟的若干基础应用和理论一、本文概述分子动力学模拟是一种基于经典力学的计算方法,通过求解分子体系的牛顿运动方程,模拟分子在特定条件下的动态行为。
该方法广泛应用于物理、化学、生物和材料科学等领域,为研究者提供了一种有效的工具,以深入理解和预测分子系统的宏观性质。
本文旨在探讨分子动力学模拟的若干基础应用和理论,从基础概念出发,阐述其基本原理、模拟方法以及在各个领域中的应用实例。
我们将详细介绍分子动力学模拟的核心技术,包括力场模型、初始条件设定、积分算法和模拟结果的解析等。
本文还将讨论分子动力学模拟的局限性以及未来的发展方向,以期为相关领域的研究人员提供有益的参考和启示。
二、分子动力学模拟的理论基础分子动力学模拟(Molecular Dynamics Simulation, MDS)是一种强大的计算技术,通过求解分子体系的牛顿运动方程,模拟分子在特定条件下的动态行为。
其理论基础主要建立在经典力学、统计力学以及量子力学之上,但在大多数应用中,由于计算能力的限制,经典力学是主要的工具。
在经典力学中,每个分子的运动可以通过牛顿第二定律来描述,即力等于质量乘以加速度(F=ma)。
在分子动力学中,这些力通常是分子间相互作用力,包括范德华力、氢键、库仑力等。
这些力可以通过分子力学模型或量子力学方法计算得出。
分子动力学模拟通常包括以下几个主要步骤:需要设定模拟的初始条件,包括分子的初始位置、速度和模拟的温度、压力等环境参数。
然后,根据分子间的相互作用力,通过求解牛顿运动方程,计算出每个分子在下一时刻的位置和速度。
这个过程会不断重复,直到模拟达到预设的时间长度或达到某种平衡状态。
在模拟过程中,为了处理大量的分子和长时间的模拟,通常会采用一些近似和简化的方法,如截断半径、周期性边界条件等。
由于分子间的相互作用力往往非常复杂,因此在模拟中通常会采用一些经验性的力场模型,如Lennard-Jones势、Morse势等。

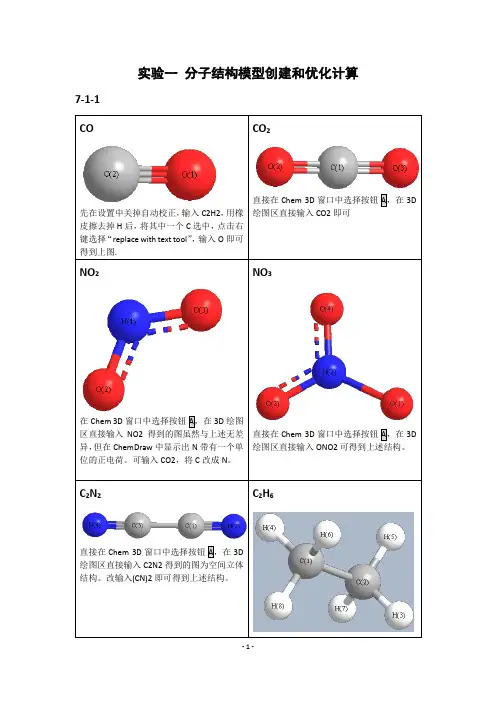
实验一分子结构模型创建和优化计算7-1-1绘图区直接输入C2H6即可。
HCOOH直接在Chem 3D窗口中选择按钮A,在3D绘图区直接输入HCOOH即可。
3DH3PO47-2-1-7-3-1 MM2O 和O 距离为2.765AE 1=-5.4331Kcal/Mol E 2= 0.0289 Kcal/Mol D=E 1-2E 2=-5.3753 Kcal/MolHF/6-31++GO 和O 距离为3.0 ÅE1= -95408.4 Kcal/mol (-152.04313 Hartrees) E2== -47701.8 Kcal/mol (-76.01774 Hartrees) D=E1-2E2=-4.8 Kcal/mol = (-0.00765 Hartrees) MP2/6-31++GO 和O 距离为2.9 ÅE1=-95650.8 Kcal/mol (-152.42932 Hartrees) E2=-47822.3 Kcal/mol (-76.20978 Hartrees) D=E1-2E2=-6.2 Kcal/mol (-0.00976 Hartrees) DFT= B3LYPO 和O 距离为2.8 ÅE1= -95870.8 Kcal/mol (-152.77997 Hartrees) E2= -47932.5 Kcal/mol (-76.38545 Hartrees) D=E1-2E2=-5.8 Kcal/mol (-0.00907 Hartrees)MOPAC-PM3O 和O 距离为3.0 ÅE1= -108.7 Kcal/molE2=-53.4 Kcal/molD=E1-2E2=-1.9 Kcal/mol7-4-2”化学意义”:RuClClPNN简介:第二代Grubbs催化剂是Grubbs在1999年对第一代催化剂的改进。
Grubbs 通过系统地对催化剂结构-性能关系进行研究,发现催化剂的活性与其中一个膦配体的解离有关,认为催化循环过程中经过一个高活性的单膦中间体,然后才与烯烃发生氧化加成。

武汉大学化学与分子科学学院《分子模拟实验》实验报告分子结构模型创建和优化计算指导老师:侯华姓名:陆文心专业:化学弘毅班学号:2012301040179日期:2014年9月25日一、实验目的无论哪种类型的计算或模拟,第一步的工作就是建模,就是创建所要研究的分子体系的三维空间模型。
这与平常所描绘的2D平面结构图不同,实际计算需要的是3D结构,每一个原子都需要三个空间坐标(x,y,z)。
因此,熟练掌握各种分子模型的创建,是计算化学最基础的技能。
实际的理论模拟计算,无论最终结果是什么,无论采用什么方法,都需要用户为所研究的分子体系提供一个合理的“初始猜测”结构,既是计算的需要,也是保证计算成功的前提之一。
当对一个新型分子体系知之甚少时,当缺乏实验数据参考时,怎么样使创建的分子模型更“合理”,也是进行本实验的目的。
本实验将介绍Chem3D提供的各种分子模型创建方法,同时介绍怎样调整、修改、显示所感兴趣的空间立体分子结构的技巧,直至满足用户的要求为止。
二、实验要求(1)熟悉Chem3D软件各项功能的含义;(2)掌握三维分子结构的创建方法;(3)掌握分子结构的调整和优化技巧;(4)了解复杂分子结构的创建和显示。
三、实验内容(1)建模:几种分子结构输入的方法;(2)处理:加氢饱和价键,加电荷;(3)优化:分子结构参数的最优化;(4)合理化:分子结构的确定。
7.1 建模的三种方式1. 分子式输入直接建模法问题7-1-1 采用该建模方法,尝试得到下列分子的正确结构:CO,CO2,NO2,NO3,C2N2,C2H6,C6H5OH,HCOOH,CH3COCH3,C2H5NO2,HNO3,H3PO4,H2CO3答:2. 化学键建模法3. 利用ChemDraw二维平面图建模4. 其它建模方式问题7-1-2 画“三键链”的结果是什么?答:结果为丙炔。
问题7-1-3 画出多个联苯环的共轭结构。
答:7.2 结构处理1. 加氢饱和价键2. 加电荷7-2-1 画出脯氨酸两种不同结构的分子骨架,并正确加氢、加电荷。
武汉大学分子模拟实验作业第十章过渡态的优化10-3-1采用GAMESS, HF/6-31G(d)计算反应物:气相:Total Energy = -313185.421886 Kcal/Mol溶剂化:Total Energy = -320855.0145 Kcal/Mol气相:Total Energy = -62343.3171 Kcal/Mol溶剂化T otal Energy =-62460.3983 Kcal/Mol反应物总能量:气相-313185.421886 Kcal/Mol+(-62343.3171 Kcal/Mol)=-375528.7389 Kcal/Mol 溶剂化后:-320855.0145 Kcal/Mol+(-62460.3983 Kcal/Mol)=-383315.4128Kcal/Mol 中间体1气相:Total Energy = -375552.4562 Kcal/Mol溶剂化:Total Energy = -544300.1875 Kcal/Mol过渡态气相:E= -375530.237194 Kcal/Mol溶剂化:E= -538290.9905 Kcal/Mol中间体2气相:E= -375611.0735 Kcal/Mol溶剂化:Total Energy = -544428.1875 Kcal/Mol 产物:气相:T otal Energy = -87245.465945 Kcal/Mol 溶剂化Total Energy = -87270.0137 Kcal/Mol气相:energy = -288356.67816 Kcal/Mol 溶剂化 energy = -288473.7594 Kcal/Mol 产物总能量:气相:-87245.465945+(-288356.67816)=-375602.1236 Kcal/Mol 溶剂化后:-87270.0137 +( -288473.7594)=-375743.7731Kcal/Mol气相反应势垒714-375570-375540BA-375528.7389-375552.4562-375530.237194-375611.0735-375602.1236F -+CH 3Cl FCH 3+Cl-中间体2中间体1TS溶剂化反应势垒0714-560000-480000-400000E /(K c a l /M o l )F -+CH 3Cl-383315.4128TS-538290.9905FCH 3+Cl--375743.7731采用Mopac,PM3方法计算气相:Total Energy = -11108.6433 Kcal/Mol溶剂化:Total Energy = -11109.9208 Kcal/Mol气相:Total Energy = -10139.2662 Kcal/Mol溶剂化:Total Energy = -10234.6847 Kcal/Mol反应物总能量:气相:-11108.6433+(-10139.2662)=-21247.9095 Kcal/Mol溶剂化后:-11109.9208+(-10234.6847)=-21344.6055 Kcal/Mol 中间体1:气相:Total Energy = -21258.0499 Kcal/Mol溶剂化:T otal Energy = -21340.9771 Kcal/Mol过渡态:气相:T otal Energy = -21252.5012 Kcal/Mol 溶剂化:Total Energy = -21320.1352 Kcal/Mol中间体2:气相:Total Energy = -21313.8820 Kcal/Mol 溶剂化:Total Energy = -21386.9799 Kcal/Mol产物:Total Energy = -13958.4182 Kcal/Mol溶剂化T otal Energy = -13959.8811 Kcal/Mol气相:Total Energy = -7348.6109 Kcal/Mol溶剂化:Total Energy = -7428.9169 Kcal/Mol 产物总能量:气相:-13958.4182+(-7348.6109)=-21307.0291 Kcal/Mol 溶剂化后:-13959.8811+(-7428.9169)=-21388.7980 Kcal/Mol气相反应势垒714-21300-21270-21240E /(K c a l /M o l )F -+CH 3Cl -21247.9095中间体1TS中间体2FCH 3+Cl--21258.0499-21252.5012 -21313.8820-21307.0291 溶剂化反应势垒714-21390-21360-21330E /(K c a l /M o l )F -+CH 3Cl TS FCH 3+Cl--21344.6055-21320.1352-21388.7980过渡态的优化1、甲酸和水分子形成的络合物的构型优化------------- GAMESS Interface ------------GAMESS Job: Minimize (Energy/Geometry) UHF/6-31G(d)Finish @ energy = -166150.964067 Kcal/Mol (-264.778634 Hartrees) ------------------------------------------2、尿素和水形成氢键------------ GAMESS Interface ------------GAMESS Job: Minimize (Energy/Geometry) UHF/6-31G(d)Finish @ energy = -188258.689979 Kcal/Mol (-300.009567 Hartrees) ------------------------------------------3、3O+2CH2OH平衡键长1.366------------ GAMESS Interface ------------GAMESS Job: Minimize (Energy/Geometry) UHF/6-31G(d)Finish @ energy = -118776.493623 Kcal/Mol (-189.282548 Hartrees) ------------------------------------------扫描----------- GAMESS Interface ------------GAMESS Job: Minimize (Energy/Geometry) UHF/6-31G(d)Finish @ energy = -46927.58803 Kcal/Mol (-74.783934 Hartrees)生成醛的过渡态:------------ GAMESS Interface ------------GAMESS Job: Optimize to Transition State UHF/6-31G(d)Finish @ energy = -118742.639222 Kcal/Mol (-189.228597 Hartrees)------------------------------------------过渡态的确定虚频为272.84 I1 2 3 4 5FREQUENCY: 272.84 I 208.13 26.24 15.82 13.97 REDUCED MASS: 10.74103 5.35183 8.27579 8.31406 8.05505 IR INTENSITY:5.18451 0.06758 0.01497 0.01009 0.034956 7 8 9 10FREQUENCY: 57.81 262.92 297.45 520.05 923.21 REDUCED MASS: 6.68774 3.17727 1.02681 4.63662 1.22834 IR INTENSITY: 0.08217 1.03754 2.85461 0.67520 0.0189411 12 13 14 15FREQUENCY: 1055.85 1157.41 1257.13 1438.30 1639.96 REDUCED MASS: 1.14502 1.25540 1.10513 1.88731 1.79969 IR INTENSITY: 0.17598 0.02378 0.44347 2.26790 1.1595016 17 18FREQUENCY: 3275.58 3389.89 3954.48REDUCED MASS: 1.05075 1.11678 1.06988IR INTENSITY: 0.11542 0.00404 0.58203心得体会:溶剂化效应(1)其相互作用:a.成键作用b.长程相互作用(2)对溶剂化效应的模拟可分为显式溶剂化模型及隐式溶剂化模型。
分子动力学模拟分子动力学是一门结合物理,数学和化学的综合技术。
分子动力学是一套分子模拟方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。
这门技术的发展进程是:1980年:恒压条件下的动力学方法(Andersenの方法、Parrinello-Rahman法)1983年:非平衡态动力学方法(Gillan and Dixon)1984年:恒温条件下的动力学方法(能势‐フーバーの方法)1985年:第一原理分子动力学法(→カー・パリネロ法)1991年:巨正则系综的分子动力学方法(Cagin and Pettit).最新的巨正则系综,即为组成系综的系统与一温度为T、化学势为μ的很大的热源、粒子源相接触,此时系统不仅同热源有能量交换,而且可以同粒子源有粒子的交换,最后达到平衡,这种系综称巨正则系综。
进行分子动力学模拟的第一步是确定起始构型,一个能量较低的起始构型是进行分子模拟的基础,一般分子的其实构型主要是来自实验数据或量子化学计算。
在确定起始构型之后要赋予构成分子的各个原子速度,这一速度是根据玻尔兹曼分布随机生成,由于速度的分布符合玻尔兹曼统计,因此在这个阶段,体系的温度是恒定的。
另外,在随机生成各个原子的运动速度之后须进行调整,使得体系总体在各个方向上的动量之和为零,即保证体系没有平动位移。
由上一步确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。
进入生产相之后体系中的分子和分子中的原子开始根据初始速度运动,可以想象其间会发生吸引、排斥乃至碰撞,这时就根据牛顿力学和预先给定的粒子间相互作用势来对各个例子的运动轨迹进行计算,在这个过程中,体系总能量不变,但分子内部势能和动能不断相互转化,从而体系的温度也不断变化,在整个过程中,体系会遍历势能面上的各个点,计算的样本正是在这个过程中抽取的。
8-1-1
NO
键长:1.1 A
键能:-81100.1 Kcal/Mol (-129.24135 Hartrees) LUMO+1 (N=8) 163.155 eV
LUMO+1 (N=7) -2.832 eV
HOMO (N=6) -2.832 eV
HOMO-1 (N=5) -12.846 eV
HOMO-2 (N=4) -21.326 eV
HOMO-3 (N=3) -21.326 eV
HOMO-4 (N=2) -23.922 eV
HOMO-5 (N=1) -52.100 eV
NO 分子轨道能级图
NO+
键长:1.0 A
键能:-80892.0 Kcal/Mol (-128.90965 Hartrees) LUMO+1 (N=8) 272.183 eV
LUMO+1 (N=7) 0.932 eV
HOMO (N=6) 0.932 eV
HOMO-1 (N=5) -11.288 eV
HOMO-2 (N=4) -22.563 eV
HOMO-3 (N=3) -22.563 eV
HOMO-4 (N=2) -24.301 eV
HOMO-5 (N=1) -54.253 eV
NO +分子轨道能级图
NO分子的总能量为:E=-2.832+2×(-12.846)+2×(-21.326)+2×(-21.326)+2×(-23.922)+2×(-52.100)=-265.872 eV
NO+分子的总能量为:E=2×(-11.521)+2×(-22.395)+2×(-22.395)+2×(-24.244)+2×(-53.973)=-269.056eV
由上述比较可以看出,NO+的分子轨道总能量低于NO分子轨道的总能量,能量越低,结构越稳定,所以NO+更稳定。
8-1-2
NO
键长:1.1318Å
键能:E= -81100.1 Kcal/Mol (-129.24135 Hartrees)
小循环:
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE ORB. GRAD INTEGRALS SKIPPED
1 0 -128.806552169 -128.806552169 0.421283477 0.000000000 28696 1
---------------START SECOND ORDER SCF---------------
2 1 -129.135589489 -0.329037320 0.381786738 0.151474544 28696 1
大循环:
1NSERCH= 1
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
N1 7.0 -0.5745456006 0.0000000000 0.0000000000
O2 8.0 0.4993950567 0.0000000000 0.0000000000
1NSERCH= 2
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
N1 7.0 -0.5932548417 0.0000000000 0.0000000000
O2 8.0 0.5181042978 0.0000000000 0.0000000000
前十个轨道的本征值:
1 2 3 4 5
-20.6943 -15.7022 -1.5838 -0.8477 -0.6473
6 7 8 9 10
-0.6274 -0.5518 -0.1053 0.0967 0.4978
其中1-8为占据轨道,9、10为未占据轨道,HOMO轨道为第8个,LUMO轨道为第9个。
NO+
键长:1.0Å
E= -80892.0 Kcal/Mol (-128.90965 Hartrees)
通过键长与能量的大小可以看出,NO的键长更长,能量较之NO+更小,稳定性高
1 2 3 4 5
-21.2344 -16.2560 -2.1937 -1.3490 -1.1712
6 7 8 9 10
-1.1712 -1.1446 -0.2704 -0.2704 0.1924
1-9为占据轨道,10为未占据轨道,HOMO轨道为第9个,LUMO轨道为第10个。
NO+键长更短,能量更低,更稳定。
CO总电子密度
键能:-70744.027355 Kcal/Mol (-112.737877 Hartrees) 键长:1.208 A
等值面值:0.002 a.u.
等值面值:0.001 a,u
等值面值:0.01 a,u
由上图可知,当等值面值小时,空间范围越大,表示分子大小和形状;当等值面值大时,空间范围越小,表示化学键信息。
8-3-1
静电势图
甘氨酸等值面:1.000a.u.
天冬酰胺等值面:1.000a.u.
静电势对总电子密度的映射图
CO分子的HOMO分子轨道对总电子密度的映射图
CO分子的LUMO轨道在总电子密度图上的映射:
UHF CALCULATION, NO. OF ALPHA ELECTRONS = 15
NO. OF BETA ELECTRONS = 14
------------ Mopac Interface ------------
Mopac Job: PM3 UHF 1SCF ALLVEC CHARGE=0 EF ESR GEO-OK GNORM=0.100 GRAPH MMOK SHIFT=80 SPIN
Spin Density:
C(1) 1.124
C(2) -0.0859
C(3) 0.0457
H(4) -0.0495
H(5) -0.0495
H(6) 0.0102
H(7) 0.0102
O(8) -0.009
O(9) 0.0015
H(10) 0.002406
Atomic Orbital Spin Density:
A. O. Density
-------- -----------
C1 s 0.154560
C1 px 0.168664
C1 py 0.077643
C1 pz 0.723121
C2 s -0.007687
C2 px -0.029087
C2 py -0.030196
C2 pz -0.018904
C3 s 0.016246
C3 px 0.005795
C3 py 0.004066
C3 pz 0.019595
H4 s -0.049535
H5 s -0.049537
H6 s 0.010203
H7 s 0.010186
O8 s 0.000020
O8 px -0.003947
O8 py -0.004156
O8 pz -0.000906
O9 s 0.000922
O9 px 0.000794
O9 py 0.000636
O9 pz -0.000902
H10 s 0.002406
Finished @ Heat of Formation = -54.70548 Kcal/Mol
自由基所在的碳原子(1)号碳上的电子云密度最大,(2)号和(3)号碳电子云密度相对来说都较小,故得出结论孤对电子没有发生离域。
8-6-1
蓝色亲水,红色疏水
氨基和羧基聚集的部位为亲水部位,脂肪烷基链聚集的部位为疏水部位
心得体会:
1. PM3为半经验公式,在计算轨道能量是会出现一定误差,导致π轨道的两个简并轨道能量差距大,如果不了解分子具体轨道构型无法判断其为π轨道。
2. 了解大小循环各自代表的意义并能在输出文件中找出来。
3. 当等值面值不同时,总电子密度图会发生改变。
当等值面值小时,空间范围越大,表示分子大小和形状;当等值面值大时,空间范围越小,表示化学键信息。
4. 对于静电视图而言,分辨率越高,图像越清晰但同时计算时间越长,因此需要正确处理两者关系,并不是分辨率越高越好。