分子模拟实验实验报告设计实验二氯卡宾
- 格式:docx
- 大小:10.53 MB
- 文档页数:6

二氯卡宾加成机理
二氯卡宾是一种活泼的有机化合物,具有很强的反应活性。
其加成机理主要包括两个步骤:首先是氯离去,形成卡宾,然后是卡宾与底物加成。
在第一步中,二氯卡宾中的氯原子在某种催化剂的作用下,从一个底物分子上脱离,形成卡宾。
这个过程需要一定的能量,通常由催化剂提供。
这一步是二氯卡宾加成反应的关键,因为此时卡宾已经形成,可以与底物分子进行加成。
在第二步中,已经形成的卡宾与底物分子进行加成反应。
根据底物的不同,卡宾可以与底物的双键、叁键或碳-氢键等发生加成反应。
加成反应的具体机制取决于底物分子的结构和卡宾的电子云分布。
通常,卡宾的电子云分布会发生变化,以便与底物分子形成共价结合。
这种电子云的重排和共价结合是加成反应的关键步骤。
二氯卡宾加成的反应机制与催化剂、底物分子和反应条件等因素有关。
在某些情况下,催化剂可以促进第一步的氯离去和第二步的加成反应。
此外,底物分子的结构和电子云分布也会影响加成反应的机制和产物。
总之,二氯卡宾的加成机理主要包括两个步骤:首先是氯离去形成卡宾,然后是卡宾与底物分子进行加成反应。
这个反应机制受到多种因素的影响,包括催化剂、底物分子和反应条件等。
了解二氯卡宾的加成机理有助于我们更好地理解其反应性质和在有机合成中的应用。

二氯双环庚烷实验报告篇一:7,7-二氯双环庚烷的制备7,7-二氯双环庚烷的制备摘要:利用季铵盐生成卡宾与不饱和键加成是一种常用的成环方法,本实验就利用二氯卡宾与环己烯反应来制备7,7-二氯双环庚烷关键词:环己烯、氯仿、7,7-二氯双环庚烷、二氯卡宾、相转移催化Abstract: The use of quaternary ammonium salts generated carbeneadducts with unsaturated bonds into the ring is a common way to prepare this experiment 7,7-dichloro-bicyclo on the use of Dichlorocarbene and cyclohexene reaction Geng alkylKeywords: cyclohexene, chloroform, 7,7-dichloro-bicyclo heptane,Dichlorocarbene, phase transfer catalysis前言:1. 二氯卡宾是有机合成中重要的中间体,与环己烯的加成反应是二氯卡宾典型反应,其产物7,7-二氯双环[ ]庚烷是重要的医药中间体.[1]2.在有机化学反应中,碳正离子、碳负离子和碳自由基是三类重要的中间体,与之对应的是亲核反应(取代和加成)、亲电反应(取代和加成)和自由基(取代和加成反应三大反应类型。
而碳自由基中,卡宾(或称为碳烯)是碳自由基中应用最广泛的。
二氯卡宾因其结构中 2 个氯原子的孤电子共轭作用,使其较为稳定,因此在卡宾中应用最多。
传统利用二氯卡宾制备三元环的方法是在无水叔丁醇中用叔丁醇钾与氯仿反应。
该方法反应时间长,产率偏低。
但加入少量相转移催化剂,则可和NaOH 溶液代替叔丁醇钾,同时大幅缩短反应时间,[2-4]提高产率。
因此,在有机化学实验教材中,常将7,7-二氯双环[]庚烷或扁桃酸的制备作为例子3. 7,7-二氯双环的庚烷的制备目前有多种制备方法,但多数都是寻早更好的相转移催化剂,如讨以环己烯和氯仿为原料,在NaOH 溶液中,用PEG-400 作为相转移催化剂合[5]成7,7-二氯双环[]庚烷可得到54%的产率.4.相转移催化剂技术目前广泛应用于各类化学中,且其应用范围在不断地扩大,在过渡金属有机催化合成中,可以利用相转移催化剂可以发挥过渡金属催化的多样性,在炭基反应,偶联[6]反应,烃化反应中有重要应用结果与讨论实验原理主反应副反应反应机理实验装置选取:二氯卡宾是非常活泼的中间体,极易与其他物质反应,因此选用了相转移催化剂TEBA来尽量避免卡宾与水的反应,同时为了使反应进行彻底和滴加NaOH溶液,和实时监控温度范围,选取了带有恒压滴液漏斗和温度计的回流装置;反应完毕后产物中有水相,有机相,也有可通过加水溶解的固体,为了分离出有机相,需要用到分液装置,同时为了将水相中残余的有机物分离出来,还要用到萃取装置;鉴于产物沸点较高的特点且高温下易分解和被氧化,选取了减压蒸馏装置来分离提纯产物.物理常数表实验现象分析:1. 由于NaOH溶解放热,溶解NaOH的过程中, 能明显感觉到杯壁变热2. 滴加NaOH一段时间后,溶液沸腾,温度计显示的温度大幅度上升,这是因为该反应是放热反应.3. 加热过程中,能看到回流现象,是由氯仿蒸汽上升遇冷凝结造成的.4. 在反应过程中,会生成HCl,而HCl进一步与NaOH反应生成NaCl,氯化钠本身在水中的溶解度不是很大,且本实验当中,整个反应体系只有9mL水,无法完全溶解生的NaCl,过剩的NaCl便以固体的形式析出来5.分液过程中,由于有机相是氯仿和产物及其他有机混在一起,因此有机相出现在下层并呈现出比水相更深的颜色产物产率误差分析及波谱分析产率误差分析:本实验产率很高,主要是氯仿造成.核磁共振氢谱由图知对应的是1,2号碳原子上的氢,对应的是3,4上面对应的氢,对应的是5,6上的氢红外光谱由图中读出:峰值和2858cm是饱和的-CH2-振动.-1峰值对应C-H变形振动-1峰值对应C-Cl伸缩振动-1-1结论:由相转移催化剂催化生成7,7-二氯双环庚烷是其普遍的生产方法,该反应虽然有活泼中间体的存在,会产生大量副产物,但比较容易除去,唯一的缺点就是无法得到高产率,从已知文献中得知不同种类的相转移催化剂对产率的提升并无太大作用.实验部分:实验仪器仪器:三口烧瓶、球形冷凝管、温度计、滴液漏斗、分液装置试剂:环己烯、TEBA、氯仿、50%NaOH、无水MgSO4、石油醚实验步骤:理论产率: 实际产量: 产率:%参考文献:[1].季铵盐A-1催化合成7,7-二氯双环庚烷.邓春雷,陈帅华,李晓如.中南工业大学学报[2].谷珉珉;贾韵仪;姚子鹏有机化学实验1991[3].兰州大学;复旦大学化学系有机化学教研室有机化学实验1994 [4].许尊乐;刘标汉有机化学实验1988[5].绿色环保催化剂PEG-400催化合成7,7-二氯双环[]庚烷的研究.林桂汕,罗轩,岑波,赖刚,韦莉华,尤诗佳.企业科技与发展2013年第10期.[6].金子林,梅建庭.温控相转移催化--水/有机两相催化新进展[J],高等学校化学报篇二:7,7-二氯双环庚烷的制备7,7-二氯双环庚烷的制备作者:xxx 学号:xxx摘要:以环己烯和三氯甲烷为原料制备7,7-二氯双环庚烷,要求了解卡宾反应制备7,7二氯双环庚烷的原理和方法,同时要求了解相转移催化的机理,在操作过程中熟练掌握减压蒸馏装置及操作方法,最后计算产物的产率。

分子模拟实验作业——设计实验-----------二氯卡宾与甲醛环加成反应的理论研究一、实验背景在大二的有机实验中,我接触到了连续合成实验,其中有一类反应为相转移催化合成·卡宾及其反应。
卡宾(Carbene)亦称为碳烯,是一类具有六个加点字的两价碳原子活性中间体,构造式为:CH2。
其中的氢原子可以被其他原子或基团所取代,这类取代物称为卡宾体或取代卡宾。
卡宾是缺电子的,具有很强的亲电性,可发生多种反应。
在有机合成中常使之与烯烃反应以制取环丙烷衍生物,上网查阅资料后,我想到了可以结合本学期所学的分子模拟理论计算方法来模拟二氯卡宾与甲醛环加成反应。
(题目来源于有机化学实验和查阅到的文献)二氯卡宾是一种取代卡宾,通常由氯仿与强碱作用产生:CHCl3 + Base :CH2 + HB + Cl-为了进一步探讨卤代卡宾与含有不对称π键物质环加成反应的机理,本文对单重态二氯卡宾与甲醛加成生成二氯环氧丙烷反应:进行了量子化学从头算的研究,得到了该反应的反应机理,并对其反应机理作了理论分析说明.同时计算了该反应在不同温度下的热力学函数和动力学性质,并作了讨论。
二、实验内容①用分子轨道理论分析二氯卡宾与甲醛环加成的机理用Chem3D软件做出二氯卡宾与甲醛的分子轨道能级图,计算分子轨道,并图示二氯卡宾和甲醛的HOMO和LUMO轨道的形状和能量。
将所有分子轨道按能级排列次序,并以此分析两反应物的轨道匹配情况。
(优化条件:Gamess Interface,HF/6-31G(d))对于该环加成反应的机理可借助于分子轨道图进行分析。
根据轨道对称匹配条件,在反应过程中,应首先是C1的2p 空轨道插入甲醛的成键π轨道,但因甲醛中的羰基是一极性基团,π键电子云密集于氧端,故C1的2p 空轨道将从氧端插入其π轨道.因π电子向C1的2p 空轨道中的迁移,从而使二者首先生成了一半环状的中间配合物。
由于二氯卡宾的ơ孤对电子与甲醛C 端的反键π轨道之间有着较强的成键作用,故随着反应的进行,二氯卡宾将在C1C20平面内按逆时针方向发生旋转.同时H1-C2和H2-C2键也由在中间配合物中与C1C20的共平面状态,通过C2-Oơ键的旋转而向着与C1C20平面垂直的方向发生转动。
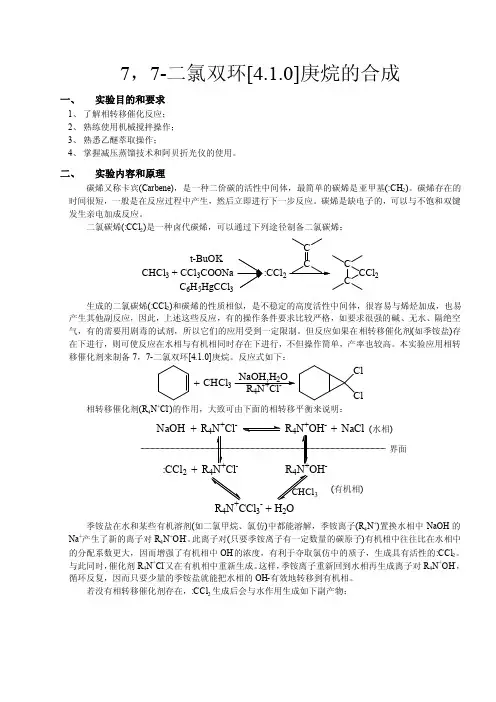
7,7-二氯双环[4.1.0]庚烷的合成一、实验目的和要求1、了解相转移催化反应;2、熟练使用机械搅拌操作;3、熟悉乙醚萃取操作;4、掌握减压蒸馏技术和阿贝折光仪的使用。
二、实验内容和原理碳烯又称卡宾(Carbene),是一种二价碳的活性中间体,最简单的碳烯是亚甲基(:CH2)。
碳烯存在的时间很短,一般是在反应过程中产生,然后立即进行下一步反应。
碳烯是缺电子的,可以与不饱和双键发生亲电加成反应。
二氯碳烯(:CCl2)是一种卤代碳烯,可以通过下列途径制备二氯碳烯:CHCl3+t-BuOKCCl3COONaC6H5HgCCl3CCCCl2CC2生成的二氯碳烯(:CCl2)和碳烯的性质相似,是不稳定的高度活性中间体,很容易与烯烃加成,也易产生其他副反应,因此,上述这些反应,有的操作条件要求比较严格,如要求很强的碱、无水、隔绝空气,有的需要用剧毒的试剂,所以它们的应用受到一定限制。
但反应如果在相转移催化剂(如季铵盐)存在下进行,则可使反应在水相与有机相同时存在下进行,不但操作简单,产率也较高。
本实验应用相转移催化剂来制备7,7-二氯双环[4.1.0]庚烷。
反应式如下:+CHCl3NaOH,H2OR4N+Cl-ClCl相转移催化剂(R4N+Cl-)的作用,大致可由下面的相转移平衡来说明:+-+-23R4N CCl3-+H2O季铵盐在水和某些有机溶剂(如二氯甲烷、氯仿)中都能溶解,季铵离子(R4N+)置换水相中NaOH的Na+产生了新的离子对R4N+OH-。
此离子对(只要季铵离子有一定数量的碳原子)有机相中往往比在水相中的分配系数更大,因而增强了有机相中OH-的浓度,有利于夺取氯仿中的质子,生成具有活性的:CCl2。
与此同时,催化剂R4N+Cl-又在有机相中重新生成。
这样,季铵离子重新回到水相再生成离子对R4N+OH-,循环反复,因而只要少量的季铵盐就能把水相的OH-有效地转移到有机相。
若没有相转移催化剂存在,:CCl2生成后会与水作用生成如下副产物:)(有机相)界面:CCl2CO +2Cl -+2H +HCOO -+2Cl -+3H +因此,在水溶液中产生出来的:CCl 2不能有效地被环己烯捕获,加成物收率很低(约为5%)。
![实验二7,7-二氯双环[4.1.0]庚烷](https://uimg.taocdn.com/48207b0afd4ffe4733687e21af45b307e871f930.webp)
实验二7,7-二氯双环[4.1.0]庚烷一.实验目的1.了解相转移催化、卡宾的生成及加成反应。
2.进一步熟悉机械搅拌装置。
3.掌握萃取、蒸馏、减压蒸馏等操作。
二.实验原理碳烯(又称卡宾carbene)是一类活性中间体总称,其通式为R2C:,最简单的卡宾是亚甲基H2C:。
卡宾存在的时间很短,一般是在反应过程中产生,然后立即进行下一步反应。
卡宾是缺电子的,可以与不饱和键发生亲电加成反应。
二氯卡宾(:CCl2)是一种卤代卡宾。
氯仿和叔丁醇作用,发生α-消除反应即得二氯卡宾。
二氯卡宾与环己烯作用,即生成7,7-二氯双环[4.1.0]庚烷:上述反应一般应在强碱而且高度无水的条件下进行。
但若利用相转移催化技术则可使反应条件温和,在水相是进行,并提高产率。
相转移催化是近十年来发展起来的一项新实验技术,对提高互不相溶两相间的反应速度、简化操作、提高产率有很好的效果。
在相转移催化剂,如季铵盐、三乙基苄基氯化铵(TEBA)存在下,氯仿与浓氢氧化钠水溶液起反应,产生的:CCl2立即与环己烯作用,生成7,7-二氯双环[4.1.0]庚烷。
一般认为该反应的机理为:在相转移催化剂存在下,在有机相中原位产生的:CCl2立即和环己烯作用,生成7,7-二氯双环[4.1.0]庚烷,产率可达60%。
为使相转移反应顺利进行,反应必须在强烈的搅拌下进行。
三.实验装置反应采用搅拌回流装置,如图3-13。
减压蒸馏装置如图2-12。
四.试剂与器材试剂:环己烯、氯仿、TEBA、氢氧化钠、乙醇、浓盐酸、无水硫酸镁。
器材:100 mL三口烧瓶、球形冷凝管、温度计、烧杯、分液漏斗、50 mL锥形瓶、搅拌马达、减压系统。
五.实验步骤在100 mL三口瓶上,依次装配好机械搅拌,回流冷凝管及温度计(图3-12)。
在三口瓶中加入10.1 mL (0.1 mol) 环己烯、30 mL (0.37mol) 氯仿和 0.5g TEBA。
将16g NaOH溶于16mL水中得到1:1的氢氧化钠水溶液。

分子植物病理学实验报告姓名:专业: 植物病理学号:实验一水稻总DNA的提取一.实验目的通过水稻叶片DNA的提取,掌握提取植物DNA的原理和方法二.实验原理十六烷基三甲基溴化铵(hexadyltrimethyl ammomum bromide,简称为CTAB), 是离子型表面活性剂,能溶解细胞膜和核膜蛋白,使核蛋白解聚,从而使DNA释放出来。
再加入氯仿等有机溶剂,能使蛋白质变性,并使抽提液分相,因核酸(DNA、RNA)水溶性很强,经离心后即可从抽提液中除去细胞碎片和大部分蛋白质。
上清液中加入无水乙醇使DNA沉淀,沉淀DNA溶于TE溶液中,即得植物总DNA溶液。
三.实验材料及仪器材料:水稻叶片(一般把种子放到培养皿中,让它长到三叶一心时即可,当然也可以从田间采集嫩的水稻叶片来提取DNA)实验方法: CTAB法试剂: 液氮,CTAB提取液, 氯仿/异戊醇(24:1),冰乙醇, 70%乙醇, 超纯水仪器:台式离心机,水浴锅,移液枪,枪头, 1.5mL或2mL离心管四.实验步骤本实验用CTAB法提取水稻基因组DNA,具体步骤如下:①将洗净的培养皿内垫上一层滤纸,将水稻种子置于滤纸上,用脱脂棉固定种子,加水保湿,将其移至光照培养箱(28℃,湿度90%,光照12小时)。
一周后即可发芽,发芽后移栽到土里,待幼苗长至三叶一心期时,即可作为提取DNA的材料。
②取三叶一心的水稻幼苗一株,将其剪碎后置于研钵中加液氮研磨成粉末状,移至已灭菌的2.0mL离心管,一般加到离心管的1/3体积。
③在离心管中加入已在65℃预热的600µL2%CTAB提取液(0.1M Tris-HCl(pH8.0),20mMEDTA(pH8.0),8%NaCl,2%十六烷基三甲基溴化铵(CTAB)),也可根据样品量适当增加提取液的量,上下颠倒混匀,置于65℃水浴锅保温30min-1h。
④将样品从水浴锅中取出,加入与提取液等体积600µL的氯仿/异戊醇(24:1),上下颠倒离心管使之充分混匀,12,000rpm离心15min。

二氯卡宾(Dichlorocarbene)是一种非常活泼的中间体,在有机化学反应中扮演着重要角色。
它的分子式为CCl2,是一种含有两个氯原子和一个碳原子的化合物。
由于其高度反应性,二氯卡宾很难以纯净形式长时间存在,通常是在反应过程中瞬间生成并立即参与反应的。
一、二氯卡宾的结构二氯卡宾的结构特点是碳原子中心带有两个氯原子,碳原子采用sp^2杂化轨道,形成一个平面三角形结构。
碳原子上还有一个未成对电子,使得二氯卡宾具有极高的反应性。
这个未配对电子使得二氯卡宾表现为一个自由基,但是由于其特殊的结构,它也可以作为一个电负性较强的亲电试剂参与反应。
二、二氯卡宾的生成方式二氯卡宾通常不是直接制备的,而是通过一些化学反应在需要时生成。
常见的生成方法包括:1. 氯仿与强碱反应:这是产生二氯卡宾最常用的方法之一。
将氯仿(CHCl3)与强碱(如氢氧化钠NaOH或氢氧化钾KOH)加热反应,可以生成二氯卡宾。
该反应的机理涉及到氯仿中的氢原子被碱去质子化,形成三氯甲烷阴离子,随后失去一个氯离子,生成二氯卡宾。
2. 卤代烃的脱卤反应:某些卤代烃在特定条件下可以通过脱卤反应生成二氯卡宾。
例如,使用锌粉作为还原剂可以将一些二卤代烃转化为相应的卡宾。
三、二氯卡宾的反应性质二氯卡宾的高反应性主要体现在以下几个方面:1. 与双键的加成反应:二氯卡宾可以与含有双键的化合物进行加成反应,生成环丙烷衍生物。
这类反应在有机合成中非常重要,可以用来合成各种环丙烷结构的化合物。
2. 插入反应:二氯卡宾能够插入到C-H键中,形成新的C-C 键。
这种反应为合成含有复杂结构的有机分子提供了一种手段。
3. 亲电性反应:由于碳原子上的未配对电子,二氯卡宾可以作为一个亲电试剂,与多种亲核试剂发生反应。
四、二氯卡宾在有机合成中的应用二氯卡宾在有机合成中的应用广泛,主要应用包括:1. 合成环丙烷衍生物:通过二氯卡宾与双键的加成反应,可以合成多种环丙烷衍生物,这类化合物在药物合成和有机材料领域具有重要应用。

无机化学实验报告,d区实验一、卡宾化反应合成碳酰基镍配合物实验目的:了解卡宾化反应中卡宾与配体的结合;通过卡宾化反应合成碳酰基镍配合物,并测定其元素分析值。
实验原理:卡宾是一类具有自由电子对的、形似中间态分子的、高度反应性的分子。
卡宾化反应是含有卡宾物质的反应,最常见的卡宾是只含碳原子的“同类卡宾”、和含有普通碳配子和卡宾的“异类卡宾”。
实验步骤:1、将固体Ni(CO)4(0.5g)放入干燥无水乙腈(30ml)中,搅拌使之溶解。
2、将三乙基胺(0.167ml)注入Ni(CO)4溶液中。
制备的NaHCO2发泡即可,外加水1 ~ 2滴。
3、将此溶液放在-78℃冰盘上,用先冷却的冰盘将甲烷化反应所需的两个溶液,分别滴入Ni(CO)4溶液中,快速搅动数分钟后,放置恢复到室温。
4、用少量5% HCl溶液用抽取器使反应混合物酸化,将混合液旋滤并洗涤,将棕黄色的沈淀洗净后放入真空干燥属于美国人。
5、测定样品的元素分析值。
实验记录与分析:合成的产物为棕黄色的沈淀,称量后得到产物为0.150g,比重为2.56/cm3。
经2次元素分析并取平均数得,实验结果如下:试剂 | 确定元素的质量/g | 称出的产物质量/g | 所含比例/ %Ni(CO)4 | Ni 29.99 | |CH2N2 | C 11.27 | 0.0425 | C 32.92计算配合物的分子式为:Ni(CO)(CH2N2)2。
实验总结:本实验进行了卡宾化反应,合成了碳酰基镍配合物Ni(CO)(CH2N2)2,并测定了其元素分析值。
实验中需要特别注意安全,如操作卡宾时需在恒温水浴中放置,同时要快速滴加试剂,酸化前要增加板滞剂避免产物损失等。
通过本实验,对卡宾化反应有了更深的了解。
![4-2【卡宾反应】7,7-二氯双环[4,1,0]庚烷的制备](https://uimg.taocdn.com/3b9c1114df80d4d8d15abe23482fb4daa58d1db2.webp)
7,7-二氯双环[4.1.0]庚烷的制备一、实验目的1.了解卡宾反应制备7,7-二氯双环[4.1.0]庚烷的原理和方法2.了解相转移催化的机理3.熟练掌握减压蒸馏装置及操作方法二、反应式+ CHCl3卡宾(carbene)是通式为R2C:的中性活性中间体的总称,具有很强的亲核性,与碳碳双键发生加成反应,生成环丙烷及其衍生物相转移催化剂(PTC):能够促使提高反应速度并在两相之间转移负离子的季铵盐和季膦盐。
这些化合物具有同时在水相和有机相溶解的能力,其中烃基是油溶性基,带电荷的氮或磷是水溶性基。
反应式中CHCl3 + HO-H2O + -CCl3,在水相中生成的-CCl3很快在相转移催化剂TEBA的作用下转入有机相,并分解成:CCl2,此中间体在有机溶剂中立即与环己烯发生加成反应,生成7,7-二氯双环[4.1.0]庚烷。
若没有相转移催化剂存在,:CCl2很快和HO-反应,几乎完全生成甲酸根离子和一氧化碳。
三、主要物料物理常数四、主要试剂及用量环己烯8.2g(10.1ml,0.1mol),氯仿44g(30ml,0.37mol),苄基三乙基氯化铵(TBAB) 0.5g,氢氧化钠五、操作步骤1.在125ml锥形瓶中,小心配制18g氢氧化钠和18ml水的溶液,冷至室温。
2.安装带搅拌、回流、温度计、滴液漏斗的反应装置。
3.在250ml的三口瓶中依次加入10.1ml环己烯、0.5gTEBA和30ml氯仿。
开动搅拌,由冷凝管上端的滴液漏斗以较慢的速度滴加配好的氢氧化钠溶液,约15min滴完。
反应物的颜色逐渐变为橙黄色。
4.滴完后,水浴中小火加热回流,保持温度在50-55℃左右,继续搅拌1h。
5.反应物冷至室温,加60ml水稀释后转入分液漏斗,分出有机层(如两界上有絮状物,可过滤),水层用25ml乙醚提取一次,合并醚层和有机层,用等体积的水洗涤两次,无水硫酸镁干燥。
6.干燥后的溶液,在水浴上蒸去乙醚和氯仿,然后进行减压蒸馏,收集产品。
![7,7-二氯双环[4.1.0]庚烷的制备](https://uimg.taocdn.com/cb7c150d59eef8c75fbfb335.webp)

7,7-二氯二环[4.1.0]庚烷的制备0一、实验目的01、通过二氯卡宾与环已烯的反应,验证二氯卡宾的存在,认识相转移催化的优越性;02、巩固搅拌器的使用、萃取、减压蒸馏等基本操作。
0二、实验原理01、二氯卡宾的制备方法02、相转移催化法0三、实验药品及其物理常数0药品名称分子量用量熔点(℃)沸点(℃)比重(d420)溶解性(水)环己烯823.2 g(0.04mol)-103.582.980.8102不溶于水氯仿119.56.5mL(0.08mol)—63.561.31.489微溶于水其它TEBA(0.2g) 50%氢氧化钠(8 mL)石油醚(40 mL) 粒状氢氧化钠(5g)无水氯化钙(2g)四、主要仪器和材料0电炉升降台电动搅拌器三口烧瓶(100mL、19#×3) 搅拌器套管玻璃搅拌球形冷凝管(19#)温度计(100℃)大小头(19#转14#) 恒压滴液漏斗空心塞水浴锅分液漏斗三角烧瓶(100mL,19#) 蒸馏装置一套, 减压蒸馏装置一套橡皮管0五、实验装置0六、实验步骤0【操作要点及注意事项】01.本反应为非均相的相转移催化反应,必须在强烈的搅拌下进行;也可用其他相转移催化剂。
02.固体溶解:可能出现泡沫乳化层,该乳化层不溶于水,也不溶于石油醚,可过滤处理。
3.分液,干燥:水层要分尽,干燥要彻底,才不会影响蒸馏;干燥要用粒状氢氧化钠。
04. 减压蒸馏:产品也可收集78-79℃/1995Pa的馏分;也可用空气冷凝管进行常压下蒸馏得到,沸程约为190-200℃。
0七、实验结果01、产品性状:;2、馏分:℃-℃、 Pa;03、理论产量:;4、实际产量:;05、产率:;0八、实验讨论01.相转移催化反应的原理。
02.氯卡宾是一种活性中间体,容易与水作用,本实验在有水存在下为什么二氯卡宾还能和烯烃发生加成反应?03.滴加氢氧化钠溶液时,强烈搅拌起何作用?0九、实验体会0谈谈实验的成败、得失。
通过卢卡斯试剂的实验,鉴别伯、仲、叔醇,了解不同类型醇与卢卡斯试剂反应的现象,从而加深对醇的分类和性质的认识。
二、实验原理卢卡斯试剂是一种无水氯化锌溶于浓盐酸的溶液,用于鉴别伯、仲、叔醇。
其原理是:醇分子中的羟基与卢卡斯试剂中的氯离子发生亲核取代反应,生成氯代烷烃和水。
叔醇、仲醇和伯醇与卢卡斯试剂反应的速度不同,叔醇反应最快,伯醇反应最慢。
三、实验仪器与试剂1. 仪器:试管、试管架、酒精灯、石棉网、滴管、烧杯、温度计、水浴锅2. 试剂:无水氯化锌、浓盐酸、乙醇、正丙醇、异丙醇、2-丁醇、苯甲醇四、实验步骤1. 分别取少量乙醇、正丙醇、异丙醇、2-丁醇、苯甲醇于五个干燥、清洁的试管中。
2. 向每个试管中加入2毫升卢卡斯试剂。
3. 将试管用木塞塞住,用力振荡,观察反应现象。
4. 将试管放入50℃的水浴中加热,观察反应现象。
五、实验结果与分析1. 乙醇与卢卡斯试剂混合后,无明显反应现象。
2. 正丙醇与卢卡斯试剂混合后,立即产生油状物,溶液分层。
3. 异丙醇与卢卡斯试剂混合后,约5分钟产生油状物,溶液分层。
4. 2-丁醇与卢卡斯试剂混合后,约10分钟产生油状物,溶液分层。
5. 苯甲醇与卢卡斯试剂混合后,立即产生油状物,溶液分层。
实验结果表明,叔醇与卢卡斯试剂反应最快,仲醇次之,伯醇反应最慢。
这与醇的反应活性有关,叔醇的碳正离子最稳定,容易生成,而伯醇的碳正离子最不稳定,难以生成。
1. 实验中,卢卡斯试剂与叔醇反应时产生油状物,说明氯代烷烃不溶于卢卡斯试剂。
2. 实验过程中,部分氯代烷烃可能挥发散失,导致反应现象不明显。
3. 在实验过程中,水浴温度对反应现象有一定影响,温度越高,反应速度越快。
4. 实验结果表明,卢卡斯试剂可以有效地鉴别伯、仲、叔醇,但在实际应用中,还需结合其他实验方法进行综合判断。
七、实验总结通过卢卡斯试剂的实验,我们了解了醇的分类和性质,掌握了卢卡斯试剂鉴别伯、仲、叔醇的方法。
实验过程中,需要注意实验条件对反应现象的影响,以及实验结果的准确性。
实验报告:二苄叉丙酮的制备与鉴定实验报告:二苄叉丙酮的制备与鉴定一、实验目的通过利用著名有机反应Claisen-Schmidt缩合反应制备二苄叉丙酮,考察有机合成、分离纯化、以及仪器分析结构表征等方面的实验技能以及解决实际问题的能力。
二、实验原理及实验内容芳香醛与含有α-氢原子的醛、酮在碱催化下能发生的羟醛缩合反应,脱水得到产率很高的α,β-不饱和醛、酮,这一类型的反应,叫做Claisen-Schmidt(克莱森-斯密特)缩合反应。
它是增长碳链的重要方法,可合成侧链上含两种官能团的芳香族化合物、以及含几个苯环的脂肪族体系中间体等。
本实验将在碱催化下,由苯甲醛和丙酮反应得到二苄叉丙酮。
二苄叉丙酮是重要的有机合成中间体,可用于合成香料、医药中间体、防日光制品等各种精细化学品。
反应方程式:苯甲醛,95%的乙醇,0.5M的氢氧化钠溶液,丙酮。
四、主要原料的物理性质名称分子式分子量熔点/℃ 沸点密度/g·cm 性状/℃178 1.0415 苯甲醛 C7H6O 106.12 -26 无色液体,具有类似苦(10/4℃) 杏仁的香味。
丙酮 C3H6O 58.08 -94.7 56.05 0.7845 无色液体,具有令人愉快的气味(辛辣甜味)。
1 / 11乙醇 C2H5OH 46.07 -114.3 78.4 0.789(158.8 (351.6K) K)318 1390 2.13 无色透明液体。
有愉快的气味和灼烧味。
易挥发。
熔融白色颗粒或条状,现常制成小片状。
易吸收空气中的水分和二氧化碳。
氢氧化钠N aOH 40.01五、实验步骤在一个装有回流冷凝管的250 ml的三颈瓶里将8.0 ml的苯甲醛溶解在80 ml 95%的乙醇中,加入80 ml 0.5M的氢氧化钠溶液和1.0 ml丙酮(用移液管量取),均匀搅拌30 min,然后用冰浴冷却,静置结晶。
通过减压过滤收集产物,用冷水洗涤。
红外箱干燥,称粗产物重量。
凝胶色谱实验报告cl 一、实验目的和要求(必填)二、实验内容和原理(必填) 三、主要仪器设备(必填)四、操作方法和实验步骤 五、实验数据记录和处理 六、实验结果与分析(必填)七、讨论、心得一、 实验目的和要求1、了解凝胶渗透色谱的测量原理,初步掌握GPC 的进样、淋洗、接收、检测等操作技术。
2、掌握分子量分布曲线的分析方法,得到样品的数均分子量、重均分子量和多分散性指数。
二、实验装置与原理 1、分离机理 GPC 是液相色谱的一个分支,其分离部件是一个以多孔性凝胶作为载体的色谱柱,凝胶的表面与内部含有大量彼此贯穿的大小不等的空洞。
色谱柱总面积Vt 由载体骨架体积Vg 、载体内部孔洞体积Vi 和载体粒间体积V0组成。
GPC 的分离机理通常用“空间排斥效应”解释。
待测聚合物试样以一定速度流经充满溶剂的色谱柱,溶质分子向填料孔洞渗透,渗透几率与分子尺寸有关,分为以下三种情况:(1)高分子尺寸大于填料所有孔洞孔径,高分子只能存在于凝胶颗粒之间的空隙中,淋洗体积Ve=V0为定值;(2)高分子尺寸小于填料所有孔洞孔径,高分子可在所有凝胶孔洞之间填充,淋洗体积Ve=V0+Vi 为定值;(3)高分子尺寸介于前两种之间,较大分子渗入孔洞的几率比较小分子渗入的几率要小,在柱内流经的路程要短,因而在柱中停留的时间也短,从而达到了分离的目的。
当聚合物溶液流经色谱柱时,较大的分子被排除在粒子的小孔之外,只能从粒子间的间隙通过,速率较快;而较小的分子可以进入粒子中的小孔,通过的速率要慢得多。
经过一定长度的色谱柱,分子根据相对分子质量被分开,相对分子质量大的在前面(即淋洗时间短),相对分子质量小的在后面(即淋洗时间长)。
自实验报告专业: 化学工程与工艺 姓名: 田小彤 学号: 3120104271 日期: 地点: 教十3009 课程名称: 高分子物理实验 指导老师: 成绩:实验名称: 汽液平衡数据的测定实验类型: 同组学生姓名:试样进柱到被淋洗出来,所接受到的淋出液总体积称为该试样的淋出体积。
分子模拟实验作业——设计实验
-----------二氯卡宾与甲醛环加成反应的理论研究
一、实验背景
在大二的有机实验中,我接触到了连续合成实验,其中有一类反应为相转移催化合成·卡宾及其反应。
卡宾(Carbene)亦称为碳烯,是一类具有六个加点字的两价碳原子活性中间体,构造式为:CH2。
其中的氢原子可以被其他原子或基团所取代,这类取代物称为卡宾体或取代卡宾。
卡宾是缺电子的,具有很强的亲电性,可发生多种反应。
在有机合成中常使之与烯烃反应以制取环丙烷衍生物,上网查阅资料后,我想到了可以结合本学期所学的分子模拟理论计算方法来模拟二氯卡宾与甲醛环加成反应。
(题目来源于有机化学实验和查阅到的文献)二氯卡宾是一种取代卡宾,通常由氯仿与强碱作用产生:
CHCl3 + Base :CH2 + HB + Cl-
为了进一步探讨卤代卡宾与含有不对称π键物质环加成反应的机理,本文对单重态二氯卡宾与甲醛加成生成二氯环氧丙烷反应:
进行了量子化学从头算的研究,得到了该反应的反应机理,并对其反应机理作了理论分析说明.同时计算了该反应在不同温度下的热力学函数和动力学性质,并作了讨论。
二、实验内容
①用分子轨道理论分析二氯卡宾与甲醛环加成的机理
用Chem3D软件做出二氯卡宾与甲醛的分子轨道能级图,计算分子轨道,并图示二氯卡宾和甲醛的HOMO和LUMO轨道的形状和能量。
将所有分子轨道按能级排列次序,并以此分析两反应物的轨道匹配情况。
(优化条件:Gamess Interface,HF/6-31G(d))
对于该环加成反应的机理可借助于分子轨道图进行分析。
根据轨道对称匹配条件,在反应过程中,应首先是C1的2p 空轨道插入甲醛的成键π轨道,但因甲醛中的羰基是一极性基团,π键电子云密集于氧端,故C1的2p 空轨道将从氧端插入其π轨道.因π电子向C1的2p 空轨道中的迁移,从而使二者首先生成了一半环状的中间配合物。
由于二氯卡宾的ơ孤对电子与甲醛C 端的反键π轨道之间有着较强的成键作用,故随着反应的进行,二氯卡宾将在C1C20平面内按逆时针方向发生旋转.同时H1-C2和H2-C2键也由在中间配合物中与C1C20的共
平面状态,通过C2-Oơ键的旋转而向着与C1C20平面垂直的方向发生转动。
随着ơ到反π授受键成键作用的不断加强,从而使中间配合物经过渡态变成了更稳定的产物二氯环氧丙烷。
图1.二氯卡宾与甲醛轨道对称性匹配图
②计算反应物,中间配合物(IC),过渡态(TS)及产物的能量和构型
在HF/6-31G(d)理论水平下优化反应物构型并计算得到能量
本文不再对中间配合物以及过渡态能量进行计算,建模时可参考下图
为了更好地观察反应途径,可以再标出二氯卡宾中的碳原子与甲醛碳原子的距离,甲醛羰基的键长。
可以观察到,在中间配合物中,C-C键逐渐变短,甲醛的C=O键长,在反应过程中逐渐拉长,故双键逐渐转化成了单键,甚至比单键更弱。
反应过程如下:
③计算反应物、中间配合物、过渡态及产物的电子结构能、零点能、总能量和相对能量。
文献值:
注:文献理论水平与本文设置的理论水平不同,计算值会有一定差异。
④绘制反应途径示意图。
根据文献结果,反应经由两步完成.第一步生成能量较低的中间配合物(IC),无能垒,且放出能量。
第二步是中间配合物异构化为产物,有能垒。
由于该能垒的数值不是很大,故第二步反应在动力学上应是较易进行的.按动力学的观点,第二步反应是总反应的速控步骤,因此,第一步反应在动力学上也应是较易进行的。
⑤计算反应的热力学性质
计算反应在不同温度下的热力学函数改变值、平衡常数K、活化熵和速率常数k。
由文献可见,在较大温度范围内,第一步反应是一个熵减少、放热而有着较大自发趋势和反应限度的反应。
第二步反应在较大的温度范围内也是一个熵减少、放热、自发趋势和反应限度较大的反应;其反应速率常数随温度的升高而明显增大,熵为负值是由于过渡态的半环状结构比中间配合物的半环状结构紧凑所致。
并由此可以确定此反应适宜发生的温度。
三、实验结论
①由于卤代卡宾在合成药物及张力小环等方面的重要价值,因而受到了许多化学家的高度重视,尤其是卤代卡宾与C=O键的加成反应.这不仅是因为该类反应的中间体一羰基叶立德一一可被用来合成人们所关心的许多目标分子,而且还由于人们对这类反应的机理了解甚少.因此,对与之相关的有重要理论意义的间题产生了进一步的研究兴趣。
然而通过卤代卡宾反应的大量实验研究可以发现,对于这类反应的机理仍限于推测水平,很有必要用量子化学的方法对其进行理论研究。
②本文利用量子化学计算方法,模拟了二氯卡宾与甲醛环加成的反应机理,反应途径,计算了相关的热力学参数与动力学参数,可以得到最佳反应温度范围。
③通过此次设计实验,使我更了解了分子模拟实验在我所学实验中的应用,体会到了计算化学独有的优势和魅力。
④还可以研究的内容:卡宾与其他物质的作用,如环己烷的生成、金属卡宾中间体的研究等等。
四、参考文献
卢巧慧,二氯卡宾与甲醛环加成反应的理论研究,物理化学学报,
Vol.14,No.9,September,1998。