分子模拟设计实验
- 格式:docx
- 大小:180.16 KB
- 文档页数:5

分子模拟的原理与实践分子模拟是指通过模拟分子之间的相互作用以及其运动状态,探究物质的性质和行为。
它是一种全面深入的研究物质结构与性质的手段,已经广泛应用于化学、生物、材料科学等诸多领域。
本文就与大家分享一下分子模拟的原理、方法及其在科学研究中的实践。
一、基本原理分子模拟的基本原理是建立分子在不同环境的各种状态下的量子力学或分子力学模型,依据这些模型来计算物质的结构、动力学和力学性质,从而得到物质性质的定量预测。
分子模型可以从两个方面考虑。
一是通过量子力学来描述分子的电子结构和原子核的运动。
二是通过分子力学来表示分子内部和分子间力的作用以及分子的构象状态和运动。
分子动力学模拟是分子模拟的一种重要方法。
它是基于牛顿力学原理和统计力学原理,模拟分子的运动和实验条件下的动力学行为,来预测它们的结构和性质。
二、模拟方法(一)分子动力学模拟分子动力学模拟是分子模拟中最为常用的方法之一。
它可以通过计算机模拟分子内部的各种物理状态,如位置、速度和位能等,在一定时间内计算出分子内部和分子间的相对位置、角度和速度等信息。
分子动力学模拟需要考虑各种参数,如能量、时间、温度等。
首先需要设置分子初始状态的坐标和速度,然后计算相互作用力和分子运动等参数,最后输出分子的位置和速度等相关信息。
(二)量子力学模拟材料和生物体系具有很强的量子效应,尤其是涉及到电子云的计算,需要使用量子力学方法进行模拟。
量子化学模拟一般使用哈密顿算符来表示能量。
通过求解薛定谔方程来计算体系的波函数,进而计算体系的电子密度和各种分子性质,如键长、键角等。
(三)平衡分子动力学模拟平衡分子动力学模拟是指使用一定温度下的分子动力学方法,模拟出物质在其中的行为和物态,从而使分子和材料结构达到动态平衡状态。
平衡分子动力学模拟可以提供有关热力学性质(如自由能、盐度等)和相对稳定性(比如液体晶体形态等)的信息。
它也可以为材料科学研究提供重要的参考依据。
三、实践案例分子模拟已经被广泛应用于生物、材料科学、纳米科技、药物研究及环境科学等领域。

分子动力学模拟实验步骤
嘿,朋友们!今天咱来聊聊分子动力学模拟实验步骤这档子事儿。
咱先得有个明确的目标吧,就好比你要去个地方,得知道去哪儿呀!这分子动力学模拟实验也一样,你得清楚自己要研究啥。
然后呢,就是选个合适的模型啦。
这就像你出门得挑双合脚的鞋子,模型不对,那后面的路可就不好走咯。
得仔细琢磨琢磨,找个能准确反映实际情况的模型。
接下来,设置好各种参数。
这可不能马虎,就跟你调电视音量似的,得恰到好处。
温度啊、压力啊、粒子间的相互作用啥的,都得考虑周全。
再之后,让计算机开始运算吧!这计算机就像个勤劳的小蜜蜂,嗡嗡嗡地帮咱干活。
咱就等着看它给出的结果。
在这过程中,你得时刻盯着点,看看有没有啥不对劲的地方。
这就好比你煮汤的时候得时不时看看火,别煮糊了呀。
等计算机算完了,就该分析结果啦。
这可需要点真本事,得从那些密密麻麻的数据里找出有用的信息。
这就像在一堆沙子里找金子,得有耐心,还得有好眼神。
分析完结果,要是不满意咋办?那就重新来呗!别灰心,科学家们不都是这样一点点摸索过来的嘛。
你说这分子动力学模拟实验像不像搭积木?一块一块地往上搭,搭错了就重新来,直到搭出你想要的那个城堡。
这中间的乐趣和挑战,只有试过才知道呀!
总之,分子动力学模拟实验可没那么简单,但也绝对不是高不可攀的。
只要咱一步一个脚印,认真去做,肯定能发现其中的奥秘。
大家加油干吧,说不定下一个重大发现就是你做出来的呢!
原创不易,请尊重原创,谢谢!。

分子动力学模拟实验的原理与方法一、引言分子动力学模拟实验是一种基于分子运动规律的计算方法,通过模拟分子间相互作用力和运动轨迹,可以研究物质的结构、性质和动力学过程。
本文将介绍分子动力学模拟实验的原理与方法,包括模拟算法、模拟体系的构建和模拟结果的分析。
二、分子动力学模拟的原理分子动力学模拟实验基于牛顿力学和统计力学的原理,通过求解分子系统的运动方程,模拟分子间相互作用力和运动轨迹。
其基本原理可以概括为以下几点:1. 分子运动方程分子动力学模拟实验中,每个分子都被看作是一个质点,其运动方程可以由牛顿第二定律得到。
根据分子的质量、受力和加速度,可以得到分子的位置和速度随时间的变化。
2. 分子间相互作用力分子间的相互作用力可以通过势能函数来描述,常见的势能函数包括Lennard-Jones势和Coulomb势。
这些势能函数描述了分子间的吸引力和排斥力,从而影响分子的相互作用和运动。
3. 温度和压力控制分子动力学模拟实验中,为了模拟实际系统的温度和压力条件,需要引入温度和压力控制算法。
常见的温度控制算法包括Berendsen热浴算法和Nosé-Hoover热浴算法,压力控制算法包括Berendsen压力控制算法和Parrinello-Rahman压力控制算法。
三、分子动力学模拟的方法分子动力学模拟实验的方法包括模拟算法、模拟体系的构建和模拟结果的分析。
下面将对这些方法进行介绍。
1. 模拟算法分子动力学模拟实验中,常用的模拟算法包括经典力场方法和量子力场方法。
经典力场方法基于经验势能函数,适用于大尺度的分子系统,如蛋白质和溶液。
量子力场方法基于量子力学原理,适用于小尺度的分子系统,如分子反应和电子结构计算。
2. 模拟体系的构建模拟体系的构建是分子动力学模拟实验中的重要步骤,包括选择模拟系统、确定初始结构和参数设置。
模拟系统的选择应根据研究的目的和问题,可以是单个分子、溶液系统或固体表面。
初始结构可以通过实验数据、计算方法或模型生成,参数设置包括力场参数、温度和压力等。

分子模拟实验报告分子光谱模拟分子光谱模拟实验报告摘要:本实验采用分子模拟的方法,通过计算机模拟的手段,研究了分子光谱。
通过构建分子模型、选择适当的计算方法和参数,得到了分子的能级结构和光谱。
实验结果表明,分子模拟可以较好地模拟分子的能级和光谱。
这种方法可以为分子光谱的研究提供一种新的途径。
1.引言分子光谱是研究分子内部能级和分子结构的重要手段。
传统的实验方法繁琐且成本较高,分子模拟则是一种新的研究手段,可以通过计算机模拟的方法得到分子的能级结构和光谱。
本实验旨在通过分子模拟的方法,研究分子的光谱现象,并探讨模拟方法的准确性和适用性。
2.实验方法2.1分子模型的构建2.2计算方法和参数的选择选择适当的计算方法和参数对于分子模拟的准确性和有效性具有重要意义。
本次实验采用量子力学方法进行计算,选择了Hartree-Fock方法作为计算方法,并设置了合适的收敛阈值和基组。
2.3能级结构的计算通过计算机程序,对构建的分子模型进行能级结构的计算。
通过求解Schrödinger方程,可以得到分子的不同能级及其能量。
2.4光谱的模拟在能级结构的基础上,模拟分子的光谱现象。
根据波长、频率和吸收强度的关系,得到分子的吸收光谱图和发射光谱图。
3.实验结果与分析3.1能级结构的计算结果通过计算机程序,得到了水分子的能级结构。
结果显示,水分子的基态电子能级为X^1A1,第一激发态能级为A^1B1、各能级的能量差异较小,符合分子光谱的特点。
3.2光谱的模拟结果根据能级结构,模拟了水分子的吸收光谱和发射光谱。
吸收光谱图显示,在不同波长范围内,水分子的吸收强度存在明显的吸收峰,这与实验观测结果一致。
发射光谱图显示,水分子在受激条件下会发出特定波长的光,这也符合实验观测结果。
4.结论通过分子模拟实验,我们成功地模拟了水分子的能级结构和光谱现象。
实验结果表明,分子模拟可以较好地模拟分子的能级和光谱,为分子光谱的研究提供了一种新的途径。

分子动力学模拟实验报告篇一:分子动力学实验报告 md2分子动力学实验报告( XX 至 XX 学年第_2_学期)班级:姓名:学号:实验名称:晶体点缺陷成绩:一、实验目的计算空位形成能和间隙原子形成能。
探究形成的空位和间隙原子所在的位置不同其形成能的变化。
以及空位和间隙原子的浓度不同时其空位能和间隙原子形成能的变化。
二、实验原理点缺陷普遍存在于晶体材料中,它是晶体中最基本的结构缺陷,对材料的物理和化学性质影响很大。
根据点缺陷相对于理想晶格位置可能出现的几种主要偏差状态,可将其命名如下:(1)空位:正常节点位置上出现的原子空缺。
(2)间隙原子(离子):指原子(离子)进入正常格点位置之间的间隙位(本文来自:小草范文网:分子动力学模拟实验报告)置。
(3)杂质原子(离子):晶体组分意外的原子进入晶格中即为杂质,杂质原子若取代晶体中正常格点位置上的原子(离子)即为置换原子(离子),也可进入正常格点位置之间的间隙位置而成为填隙的杂质原子(离子)。
一般情况下,空位、间隙原子都是构成晶体的原子或离子偏离原有格点所形成的热缺陷。
在一定温度下,晶体中各原子的热振动状态和能量并不同,遵循麦克斯韦分布规律。
热振动的原子某一瞬间可能获得较大的能量,这些较高能量的原子可以挣脱周围质点的作用而离开平衡位置,进入到晶格内的其他位置,于是在原来的平衡格点位置上留下空位。
根据原子进入晶格内的不同位置,可以将缺陷分为弗伦克尔(Frenkel)缺陷和肖特基(Schottky)缺陷。
点缺陷都只有一个原子大小的尺度,因此不容易通过实验对其进行直接的观察。
而且实验方法研究缺陷时利用较多的还是缺陷对晶体性质的影响。
例如,通过测量晶体的膨胀率和电阻率的变化规律,即可对点缺陷的存在、运动和相互作用等方面展开间接的研究。
分子动力学方法对金属材料原子尺度物理和化学过程的研究具有实验法无法比拟的优势,可直观的模拟和分析晶体中的点缺陷。
若我们搭建完整晶体的原子个数为N,能量为E1,通过删除和增加一个原子得到空位和间隙原子,充分弛豫后体系能量为E2,则空位形成能Ev 和间隙原子形成能Ei分别为:三、实验过程(1)进入2_point文件夹$cd口2_point(2)运行in.inter文件,得到Cu的八面体间隙原子的图像,以及体系的总能量的变化,计算出八面体间隙原子的形成能。

分子动力学的模拟程序设计
一、简介
分子动力学模拟是一种利用数值模拟技术和分子结构理论来模拟构成
分子系统的静态和动态性质的方法,它是应用微观热力学和分子力学理论,通过计算每个粒子(原子集合)之间的相互作用力系数而对原子、分子等
分子系统的历史运动进行数学模拟,从而计算系统的物理性质及其反应,
实现对系统的理论预测,是计算物理学和化学研究的重要方法,已经成为
科研、工程设计领域不可缺少的工具。
1.建立模拟系统:首先要构建模拟系统。
实验室中可以准备一些模板,例如金刚石、碳纳米管等,可以通过计算机将模板转化为经典分子动
力学的模拟系统,也可以利用一些已有的软件,如VMD(可视化分子动力学)等来构建模拟系统。
2.建立分子动力学模型:根据需要检测的性质,确定所采用的分子
动力学模型,辅以计算细节,比如时间步长,以及模拟的温度和压强等,
以及对运动量的护航或管理。
3.模拟系统参数输入:确定系统的模拟参数,输入到程序中。
4.模拟系统进行演化:然后就可以开始对系统进行演化仿真模拟,
在这个过程中,可以计算各种性质。

使用Blender进行分子模拟:分子动力学实验Blender是一款强大的开源三维计算机图形软件,除了在电影和游戏制作中得到广泛应用外,它还可以用于分子模拟。
分子模拟是一种研究分子行为的重要方法,通过模拟分子在不同环境下的运动轨迹和相互作用,可以揭示许多生物和物理现象的基本原理。
在本教程中,我们将介绍如何使用Blender进行分子动力学实验。
首先,我们需要准备好所需的材料。
在进行分子模拟之前,我们需要从科学文献或其他来源获取分子的结构信息,包括原子坐标、键长、键角和二面角等。
一旦我们有了这些信息,我们可以使用Blender的“分子建模”工具将分子结构导入到软件中。
接下来,我们需要设置分子的力场参数。
力场参数是描述分子相互作用的一组规则和方程式,可以模拟原子之间的化学键和非共价相互作用。
Blender提供了许多常用的力场参数,可以根据不同的分子类型进行选择。
我们可以在Blender的选项面板中设置这些参数,以便更准确地模拟分子的行为。
然后,我们需要设置分子的模拟条件。
这包括温度、压力和模拟时间等参数。
在模拟过程中,我们可以通过改变这些参数来研究分子在不同环境下的行为。
Blender提供了一个直观的界面,可以轻松地调节这些参数,并观察分子在不同条件下的运动和相互作用。
现在,我们可以开始进行分子动力学实验了。
分子动力学实验是通过求解牛顿力学方程来模拟分子的运动和相互作用。
在Blender中,我们可以使用其内置的分子动力学引擎来实现这一过程。
通过将分子放置在一个模拟单元中,应用适当的力场参数和模拟条件,我们可以模拟分子在任意时间段内的行为。
Blender还提供了一系列分析工具,可以用于评估模拟结果,包括分子动力学轨迹、能量变化和分子结构的变化等。
最后,在完成分子动力学实验后,我们可以通过Blender的可视化功能来展示分子的运动轨迹和相互作用。
Blender的渲染功能可以将分子的三维结构以及模拟结果呈现为逼真的图像和动画。

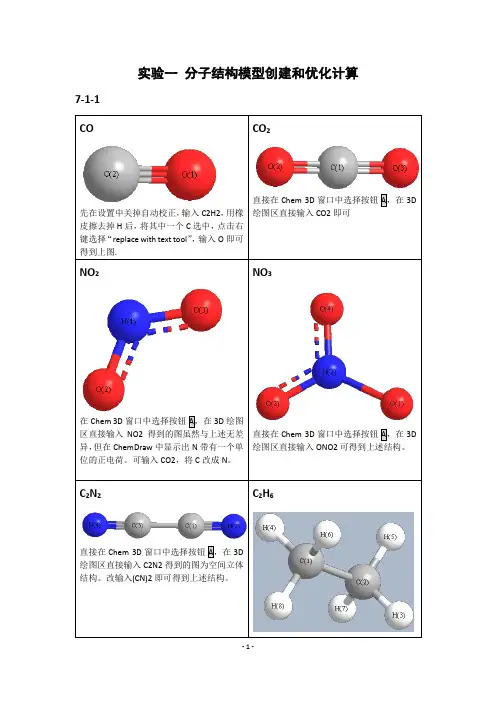
实验一分子结构模型创建和优化计算7-1-1绘图区直接输入C2H6即可。
HCOOH直接在Chem 3D窗口中选择按钮A,在3D绘图区直接输入HCOOH即可。
3DH3PO47-2-1-7-3-1 MM2O 和O 距离为2.765AE 1=-5.4331Kcal/Mol E 2= 0.0289 Kcal/Mol D=E 1-2E 2=-5.3753 Kcal/MolHF/6-31++GO 和O 距离为3.0 ÅE1= -95408.4 Kcal/mol (-152.04313 Hartrees) E2== -47701.8 Kcal/mol (-76.01774 Hartrees) D=E1-2E2=-4.8 Kcal/mol = (-0.00765 Hartrees) MP2/6-31++GO 和O 距离为2.9 ÅE1=-95650.8 Kcal/mol (-152.42932 Hartrees) E2=-47822.3 Kcal/mol (-76.20978 Hartrees) D=E1-2E2=-6.2 Kcal/mol (-0.00976 Hartrees) DFT= B3LYPO 和O 距离为2.8 ÅE1= -95870.8 Kcal/mol (-152.77997 Hartrees) E2= -47932.5 Kcal/mol (-76.38545 Hartrees) D=E1-2E2=-5.8 Kcal/mol (-0.00907 Hartrees)MOPAC-PM3O 和O 距离为3.0 ÅE1= -108.7 Kcal/molE2=-53.4 Kcal/molD=E1-2E2=-1.9 Kcal/mol7-4-2”化学意义”:RuClClPNN简介:第二代Grubbs催化剂是Grubbs在1999年对第一代催化剂的改进。
Grubbs 通过系统地对催化剂结构-性能关系进行研究,发现催化剂的活性与其中一个膦配体的解离有关,认为催化循环过程中经过一个高活性的单膦中间体,然后才与烯烃发生氧化加成。
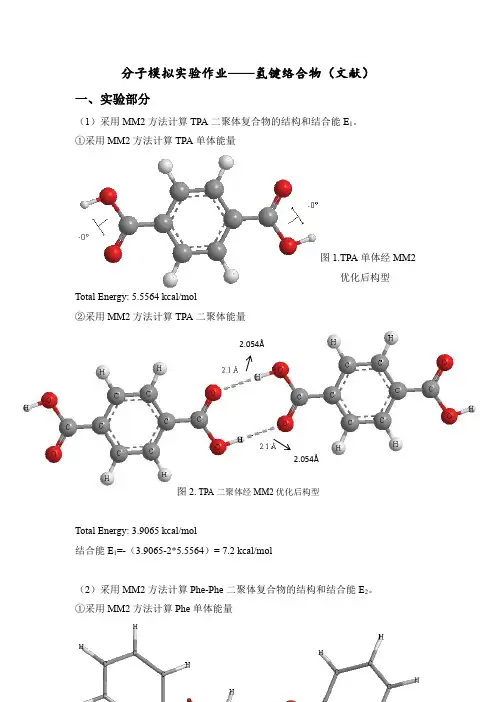
分子模拟实验作业——氢键络合物(文献)一、实验部分(1)采用MM2方法计算TPA 二聚体复合物的结构和结合能E 1。
①采用MM2方法计算TPA 单体能量图1.TPA 单体经MM2优化后构型Total Energy: 5.5564 kcal/mol②采用MM2方法计算TPA 二聚体能量图2. TPA 二聚体经MM2优化后构型Total Energy: 3.9065 kcal/mol结合能E 1=-(3.9065-2*5.5564)= 7.2 kcal/mol(2)采用MM2方法计算Phe-Phe 二聚体复合物的结构和结合能E 2。
①采用MM2方法计算Phe 单体能量2.054Å2.054Å图3. Phe-Phe单体经MM2优化后构型Total Energy: -2.6281 kcal/mol②采用MM2方法计算TPA二聚体能量(第一种)2.015Å2.496Å图4. Phe-Phe二聚体经MM2优化后构型Total Energy: -23.3587 kcal/mol结合能E2= -(-23.3587 -(-2.6281*2))= 18.1 kcal/mol③采用MM2方法计算TPA二聚体能量(第二种)2.073Å图5. Phe-Phe二聚体经MM2优化后构型Total Energy: -27.9305 kcal/mol结合能E2= -(-27.9305 -(-2.6281*2))= 22.7 kcal/mol(3)采用MM2方法计算Phe-Phe-TPA二聚体复合物的结构和结合能E3。
通过结合能的比较,验证与实验结果E2>E3>E1是否符合?①采用MM2方法计算Phe-Phe-TPA二聚体复合物2.071Å2.011Å图6. Phe-Phe-TPA二聚体经MM2优化后构型Total Energy: -7.0116 kcal/mol结合能E3= -(-7.0116-5.5564 -(-2.6281))= 10.0 kcal/molE1= 7.2 kcal/molE 2= 15.8 /22.7kcal/mol E 3= 10.0 kcal/mol15.8>10.0>7.2 即E 2>E 3>E 1 与实验结果E 2>E 3>E 1相符合(4)设计出如图2所示的2TPA-2Phe 四聚体复合物的结构并计算其结合能。

分子动力学模拟实验注意事项分子动力学模拟实验是一种重要的科学研究方法,它可以通过模拟分子的运动和相互作用,揭示物质的性质和行为规律。
然而,要进行一次成功的分子动力学模拟实验,并不是一件容易的事情。
下面,我将介绍一些分子动力学模拟实验的注意事项,希望对研究者们在实验中能够有所帮助。
首先,选择适当的模拟软件是非常重要的。
目前市场上有许多分子动力学模拟软件可供选择,如GROMACS、LAMMPS等。
在选择软件时,需要考虑实验所需的功能、计算速度、用户友好性等因素。
同时,还要注意软件的稳定性和可靠性,选择经过验证和广泛应用的软件,以确保实验结果的可信度。
其次,准备好适当的模型和初始结构是进行分子动力学模拟实验的基础。
模型的构建需要根据研究的目的和所研究的物质特性进行合理设计。
在构建模型时,需要考虑分子的拓扑结构、键长、键角、电荷分布等因素,并根据实验需求添加适当的边界条件和约束条件。
初始结构的选择也非常重要,需要根据实验的目标和所研究的系统特性进行合理选择,以确保模拟结果的准确性和可靠性。
在进行分子动力学模拟实验时,还需要注意选择合适的力场和模拟参数。
力场是描述分子间相互作用和运动规律的数学模型,不同的力场适用于不同类型的物质系统。
选择合适的力场可以提高模拟结果的准确性和可信度。
同时,还需要选择合适的模拟参数,如温度、压力、时间步长等。
这些参数的选择需要根据实验的目标和所研究的系统特性进行合理调整,以确保模拟结果的可靠性和可重复性。
此外,在进行分子动力学模拟实验时,还需要考虑系统的尺寸和边界条件的设置。
系统的尺寸需要根据实验的目标和所研究的物质特性进行合理选择,以确保模拟结果的准确性和可靠性。
边界条件的设置也非常重要,可以通过周期性边界条件或壁约束等方式来模拟无限大系统或受限系统,以更好地反映实际情况。
最后,分子动力学模拟实验的结果分析和解释也是非常重要的。
实验结果的分析需要根据实验的目标和所研究的系统特性进行合理选择,可以通过计算分子的平均位置、速度、能量等物理量来了解系统的性质和行为规律。
分子模型操作实验报告分子模型操作实验报告摘要:本实验旨在通过分子模型操作,深入了解分子结构和化学键的形成。
通过实验操作,我们对分子模型的构建和分子间相互作用有了更深入的理解,并通过实验结果验证了一些化学原理。
引言:分子模型是一种用于可视化分子结构的工具,通过模拟真实分子的形状和键的连接方式,帮助我们更好地理解分子间的相互作用和化学反应。
本实验中,我们使用塑料球和棍子构建分子模型,并通过操作模型来观察分子的结构和性质。
实验步骤:1. 准备工作:准备所需的塑料球和棍子,根据实验需要选择不同颜色的球和棍子,以区分不同原子和键的类型。
2. 构建分子模型:根据给定的化学式或分子结构,使用球和棍子按照正确的键连接方式构建分子模型。
3. 观察分子结构:观察分子模型的形状、原子间的距离和键的角度,了解分子的空间构型和立体结构。
4. 操作分子模型:通过操作分子模型,改变原子的位置或键的角度,观察分子结构的变化和对应的性质变化。
5. 模拟化学反应:根据给定的化学反应方程式,通过改变分子模型中原子的位置和键的连接方式,模拟化学反应的进行过程。
6. 分析实验结果:根据实验观察和模拟化学反应的结果,分析分子结构和化学键对分子性质的影响。
实验结果和讨论:通过实验操作和观察,我们发现分子的形状和结构对其性质具有重要影响。
例如,分子中原子的位置和键的角度改变,可能导致分子的立体异构体形成,从而影响分子的化学活性和反应性。
此外,我们还发现不同类型的化学键(如共价键和离子键)对分子的稳定性和物理性质有不同的影响。
在模拟化学反应过程中,我们观察到分子模型中原子的位置和键的连接方式的改变,可以模拟化学反应中的键的形成和断裂过程。
通过这种方式,我们可以更直观地理解化学反应的机理和过程,并预测反应的产物和副产物。
结论:通过分子模型操作实验,我们深入了解了分子结构和化学键的形成。
通过实验操作和观察,我们对分子的形状和结构对其性质的影响有了更深入的理解。
分⼦模拟实验综合实验分⼦模拟实验作业——综合实验⼀、实验部分1.反应物和产物分⼦的电⼦结构(1)画出反应物和产物分⼦的⽴体结构图,分别⽤“wireframe(线条型)”,“ball & stick(球棍型)”,“cylindrical bonds(键线型)”,“space filing(实⼼球型)”(3)计算分⼦轨道,并图⽰各个分⼦的HOMO和LUMO轨道的形状和能量。
将所有的分⼦轨道按能级排列次序,并以此分析两反应物的轨道匹配情况。
C4H6LUMO HOMOC2H4LUMO HOMOC6H10LUMO HOMO反应是由1,3-丁⼆烯的HOMO与⼄烯的LUMO反应,故图为分⼦轨道很匹配,有利于加成反应。
DA反应的反应物分为两部分,双烯体提供共轭双烯,亲双烯体提供不饱和键。
DA反应是由双烯体的HOMO与亲双烯体的LUMO发⽣作⽤,反应过程中,电⼦从双烯体的HOMO流⼊亲双烯体的LUMO。
C2H4的LUMO轨道与C4H6的HOMO轨道电⼦云相匹配,正好符合双烯合成的条件,利于反应的进⾏。
(4)绘制反应物和产物分⼦的总电⼦密度图和静电势图,分析两个反应物分⼦的电性匹配情况。
总电⼦密度图静电势图总电⼦密度图静电势图由轨道匹配来说,要使得反应能够顺利进⾏,必须是同为正或负。
C2H4的静电势图与C4H6的静电势,很匹配,所以有利于加成反应。
2.构象搜索与分⼦间长程相互作⽤(1)计算丁⼆烯分⼦绕CCCC⼆⾯⾓转动的构象,确定稳定构型,并计算内转动的能垒⾼度。
(可能不⽌⼀个)此时的构型有两种,顺式构型和反式构型。
稳定构型为反式构型,内转动能垒⾼度:40.58kcal/mol(反式构型)和38.67kcal/mol(顺式构型)。
图1 扫描势能⾯时的构型能量单位:kcal/mol①势能⾯图:图2丁⼆烯分⼦绕CCCC ⼆⾯⾓转动的构象②相互作⽤势能线及拟合图选取每个距离下的最低能量,减去MM2优化得到的C2H4和C4H6的能量 C2H4:Total: 0.4267 C4H6:Total: -0.1615 数据表格如下:CCCC ⼆⾯⾓/(°)质⼼距离/?质⼼距离/?相互作⽤能量(kc al/图3 C 4H 6与C 2H 4相互作⽤势能曲线3 反应途径计算(1)在HF/6-31G(d)理论⽔平上计算两个反应物分⼦和产物分⼦的298.15K时标准⽣成焓。
新型药物物质的分子模拟研究随着人类对药物研究的深入,传统的试错研究方法已经不能满足现代药物研究的需求。
因此,分子模拟技术逐渐成为药物研究的重要工具之一。
新型药物物质的分子模拟研究,已经被广泛应用于药物发现、理解药效与毒性基础机制等方面。
一、分子模拟技术分子模拟技术是一种计算机辅助的化学研究方法,通过计算机运算模拟分子体系的结构和性质,以解决实验难以探测或无法确定的化学问题。
分子模拟技术的实现需要多个步骤。
首先,需要确定所要处理的分子体系,并收集关于该体系的实验数据。
然后,需要对该分子体系进行建模,以确定其几何结构和必需的物理化学参数。
接下来,使用计算机运行相关算法,模拟该分子体系的动力学运动,并分析其一系列性质和反应过程,以预测药效和毒性等关键性能参数。
二、分子模拟在药物研究中的应用1.药物发现分子模拟技术在药物发现中有着重要的应用。
通过分子模拟技术,我们可以预测某些化合物的结构和性质,从而为药物设计和发现提供有价值的信息。
例如,可以在药物设计的早期阶段,使用分子模拟技术评估化合物的合适性。
通过模拟分析化合物的分子结构、特性、相互作用等方面,从而预测其生物活性,提前排除可能无效或有害的候选分子。
同时,针对分子药物、天然药物、蛋白药和小分子化合物药等类型的化合物,可以使用分子动力学模拟验证其适性。
2.药物毒性研究分子模拟技术也可以用于药物毒性研究。
实验室动物或临床试验前需要评估药物的毒性,一般需要进行多个试验和分析,而这些试验费用昂贵且耗时长。
通过分子模拟技术,可以预测药物对生物体系的毒性,帮助研究人员更快速、更准确地了解药物的危险性和其潜在副作用。
例如,可以通过在分子水平上分析某种分子的构象和相互作用能,从而预测其的化学中毒性机制、可达毒性程度和相关的慢性毒性程度等。
3.强大的计算能力分子模拟技术本身就有着强大的计算能力,因此可以简化诸如量子机械研究或其他计算化学方法的计算问题。
例如,可以通过量子计算方法,和分子错位之间和分子形状的不规整性之间的相互作用等因素,通过几何优化、热力学分析、傅立叶变换等方法来预测分子的动态和反应特性。
分子动力学模拟实验的原理和应用分子动力学模拟实验是一种利用数学和计算机模型来研究分子运动规律和相互作用的方法。
它被广泛应用于物理、化学、材料科学、生物化学等领域,为人类探索物质世界提供了重要的工具。
下面我们将探讨这种方法的原理和应用。
一、分子动力学模拟实验的原理分子动力学(Molecular Dynamics, MD)是一种基础的计算物理学方法,它使用牛顿运动定律和量子力学原理,将原子和分子的运动看作是经典粒子在势能场中的运动。
通过将势能函数数值化为分子内原子之间的相互作用,将分子所受的力的大小和方向计算出来,并根据牛顿运动定律来确定它们的轨迹和状态。
这样可以得到分子在不同时间点的位置、速度、能量等信息,进而研究其热力学、动力学和结构性质。
MD模拟计算主要分为以下几个步骤:首先定义分子体系,包括原子种类、原子数、体系大小、温度、压力等参数;然后定义分子力场,包括势能和力的计算方法;根据分子力场计算出分子所受的力;根据牛顿运动定律求解分子在不同时间点的位置和速度;最后计算分子的热力学、动力学和结构性质。
二、分子动力学模拟实验的应用MD模拟是一种基于物理原理的理论模型,可以模拟不同温度、压力、相变等条件下的分子运动和相互作用。
它可以为化学反应、材料合成、酶催化机理、药物设计等研究提供重要的帮助。
以下是MD模拟在不同领域的应用。
1. 材料科学MD模拟可以模拟材料的物理、化学性质及其相互作用。
例如,在研究聚合物和复合材料的合成、结晶、玻璃转变和热机械性能时,MD模拟可计算热力学、动力学参数和结构特征,并预测材料的制备和性能。
2. 生命科学MD模拟常用于分析生物大分子的结构、动力学和解析度。
例如,在研究蛋白质折叠、膜蛋白通道和酶促反应中,可以通过模拟蛋白质水合、静电作用和氢键的形成,从而探索蛋白质分子结构和功能等生物学问题。
3. 药学MD模拟可用于研究药物的作用机制、药物相互作用和药效等问题。
例如,在研究药物与细胞膜接触时,可以通过模拟药物与膜蛋白的相互作用,预测药物与载体的相互作用、吸收性和药效。
分子模拟实验作业——生物大分子一、实验部分12-3-1获得PDB 号为“1HCK”的蛋白(human -cyclin -dependent kinase 2,i,e.,CKD2和ATP 的结合晶体结构),并采用不同的模型观察其特点①分别用卡通模型和丝带模型显示生物大分子结构,并用球棍模型、棒状模型显示其中小分子、金属离子等。
丝带模型&球棍模型卡通模型&棒状模型参考文献: Analysis of CDK2 Active-SiteHydration: A Method to DesignNew Inhibitors Zdeneˇk Krˇı´zPROTEINS: Structure, Function,and Bioinformatics 55:258–274(2004)12.2 分子对接①聚合物对接前效果图GLU81 1.9ÅASP86 2.1ÅLYS 33 1.9Å②聚合物对接后效果图对接后实际距离和设置的最优值12-3-2在样本文件中,创建冰的晶体结构,分别做温度为260K,273K,298K,373K下的分子动力学模拟(10 ps),观察晶体机构的变化情况,并做定性解释。
①不同温度下冰晶体结构图:原始冰晶体结构图由冰晶体在不同温度下的结构可见,随温度升高,冰晶体的各个水分子之间的距离不断增加,晶体结构趋向于分散无序状。
②不同温度下,冰晶体分子动力学模拟图③不同温度下体系的总能量与势能由曲线形状可见,经过分子动力学模拟之后,体系的能量降低,变得更加稳定。
由计算结果可见,体系的总能量和势能随温度的升高而增大。
因为当温度升高时,分子的热运动加剧,使分子的伸缩、转动、振动势能增加从而使分子总能量增加,而体系的是能增加是因为非键相互作用尤其是分子间氢键相互作用减弱。
二、实验心得与体会本次实验主要进行了生物大分子的模拟。
生物大分子一般包含上千个原子,目前还不能应用量子化学从头计算方法模拟,常用的方法有QM/MM方法,和纯粹的分子动力学模型。
分子模拟实验设计实验
杨平
化基一班
2012301040010
星期三下午
指导老师:侯华
利用量子化学知识模拟Ritter reaction的机理
背景
Ritter反应,即腈类和容易形成碳正离子的化合物,诸如烯、醇、羧酸、酯、酮等在强酸存在的条件下发生的一类反应,该反应能生成N-取代的酰胺或胺类化合物(将N-取代的酰胺水解即得),是构筑C-N键最为重要的方法之一。
Ritter反应是一类重要的有机合成反应,可通过烯烃或醇与腈的直接反应制备酰胺。
该反应不但具有原子经济性,而且有很好的应用前景。
本反应广泛应用于精细有机合成中,包括药品、农药、高分子行业用的功能单体的合成,诸如高分子功能单体N- 异丙基丙烯酰胺的合成。
所以弄清楚该反应的机理显得十分有必要。
而经过一学期分子模拟实验的学习,特别是化学反应模拟章节的学习让我对通过Chem3D软件模拟反应过渡态从而得出反应机理有了初步了解。
Chem3D软件模拟是一种很好的模拟反应进行过渡态及中间体的方法。
当然对于Ritter reaction的机理研究已经非常成熟,下图为反应的机理图:
首先形成碳正离子,任何能够形成稳定碳正离子的反应物都可以成为起始原料。
然后碳正离子进攻氰基氮原子,生成的正离子迅速加水,转变为N- 烃基取代酰胺。
还有相似的反应
实验部分
下面用分子模拟实验课堂上学到的相关化学反应模拟的知识来进行模拟。
之所以想用Chem3D进行模拟计算,是因为通过模拟计算可以从能量的角度出发来更好地理解Ritter reaction.
1.分子结构优化
对分子结构的优化采用HF/6-31G(d)基组,计算出反应物与产物的能量
叔丁醇
乙腈
酰胺
2.根据反应的机理图,得到反应中间体,并计算能量(HF/6-31G(d))
(1)碳正离子的形成
(2)
(3)水进攻叁键碳原子,经过质子转移即得N-取代酰胺
3.通过计算的能量得到反应能量途径
通过origin9.0软件做出反应的能量途径
由于在个人电脑上安装的Chem3D软件很多功能用不了,用HF/6-31G(d)计算出的能量也与实验室机房中计算的有很大出入,所以所有关于实验数据部分
的内容均没有附上,敬请老师原谅。
实验结论
通过利用Chem3D和origin软件计算有关Ritter reaction中的能量变化,我们可以总结出该反应大概有以下几步组成。
以叔醇为例,①叔醇在强酸性溶液中生成稳定的三级碳正离子,②该碳正离子受到腈氮原子的亲核进攻,生成一个腈鎓离子(Nitrilium ion)。
③第一步中生成的水进攻叁键碳原子,经过质子转移即得N-取代酰胺,水解可以得胺。
实验感悟
通过对Ritter reaction用量子化学的方法进行模拟,通过比较能量大小这种非常直观的方法可以帮助我们更好地理解和掌握该反应。
为我们以后使用该反应打下良好的基础。
当然我们也还可以用Chem3D软件对该反应过程进行更加详细的描述。
例如可以通过改变两反应物分子之间的距离来计算不同距离时的能量值,通过势能曲线的绘制得到能量最低时两者之间的距离,并计算出势垒高度;还可以通过Mopac中的COSMO溶剂模型,计算该反应在一些有机溶剂中的反应热和活化能,并分析溶剂是否对该反应有利。
总之,通过分子模拟的辅助可以让我们对一个反应有更深更好的理解和掌握,将对我们下一阶段的学习研究打下坚实的基础。
参考文献
1. (a) Ritter, J. J.; Minieri, P. P. J. Am. Chem. Soc. 1948, 70, 4045−4048.
(b) Ritter, J. J.; Kalish, J. J. Am. Chem. Soc. 1948, 70, 4048−4050.
2. Krimen, L. I.; Cota, D. . React. 1969, 17, 213–329. (Review).
3. Top, S.; Jaouen, G. J. Org. Chem.1981, 46, 78−82.
4. Jie Jack Li.《Name Reaction》.2010,486-487.。