酮基布洛芬生产工艺研究与改进
- 格式:pdf
- 大小:698.64 KB
- 文档页数:3


第1篇一、实验目的本实验旨在通过化学合成方法制备布洛芬,并对其合成过程进行详细记录,以了解布洛芬的合成工艺及反应条件。
二、实验原理布洛芬(Ibuprofen)是一种非甾体抗炎药(NSAID),具有解热、镇痛、抗炎作用。
本实验采用芳基1,2-转位重排法合成布洛芬,以异丁基苯为起始原料,经过傅克反应、催化加氢、氧化、酸催化等步骤,最终得到目标产物。
三、实验材料与仪器1. 实验材料:(1)异丁基苯:分析纯(2)三氯化铝:分析纯(3)铁粉:分析纯(4)硝酸:分析纯(5)氢氧化钠:分析纯(6)无水乙醇:分析纯(7)水合肼:分析纯(8)硝酸银:分析纯2. 实验仪器:(1)圆底烧瓶(2)分液漏斗(3)滴液漏斗(4)冷凝管(5)锥形瓶(6)烧杯(7)抽滤瓶(8)干燥器(9)紫外-可见分光光度计(10)红外光谱仪四、实验步骤1. 傅克反应:将50 mL异丁基苯、10 mL三氯化铝和20 mL无水乙醇混合,加入三氯化铝,搅拌溶解。
然后将混合液滴加到装有20 mL三氯化铝和30 mL无水乙醇的圆底烧瓶中,加热回流1小时。
2. 催化加氢:将傅克反应得到的产物冷却至室温,过滤,用无水乙醇洗涤,干燥。
将得到的固体加入装有50 mL无水乙醇的圆底烧瓶中,加入5 g铁粉,加热回流3小时。
3. 氧化:将催化加氢得到的产物冷却至室温,过滤,用无水乙醇洗涤,干燥。
将得到的固体加入装有50 mL硝酸和20 mL水的烧杯中,加热回流2小时。
4. 酸催化:将氧化得到的产物冷却至室温,过滤,用无水乙醇洗涤,干燥。
将得到的固体加入装有50 mL水合肼和20 mL硝酸的烧杯中,加热回流2小时。
5. 纯化:将酸催化得到的产物冷却至室温,过滤,用无水乙醇洗涤,干燥。
将得到的固体加入装有50 mL硝酸银和20 mL水的烧杯中,加热回流2小时。
6. 结晶:将纯化得到的产物加入装有100 mL无水乙醇的烧杯中,冷却至室温,抽滤,干燥,得到布洛芬。
五、实验结果与分析1. 反应条件对产率的影响:(1)三氯化铝的用量:当三氯化铝用量从5 mL增加到10 mL时,产率从40%提高到60%。
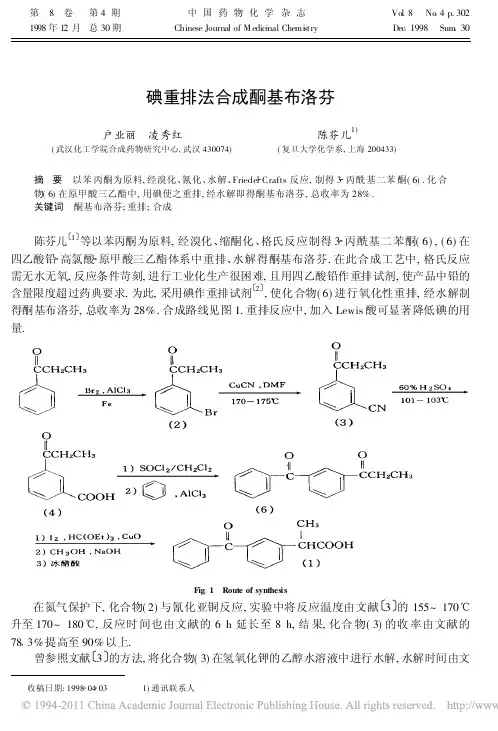
收稿日期:1998 04 03 1)通讯联系人碘重排法合成酮基布洛芬户业丽 凌秀红(武汉化工学院合成药物研究中心,武汉430074) 陈芬儿1)(复旦大学化学系,上海200433)摘 要 以苯丙酮为原料,经溴化、氰化、水解、Friedel Crafts 反应,制得3 丙酰基二苯酮(6).化合物(6)在原甲酸三乙酯中,用碘使之重排,经水解即得酮基布洛芬,总收率为28%.关键词 酮基布洛芬;重排;合成陈芬儿 1 等以苯丙酮为原料,经溴化、缩酮化、格氏反应制得3 丙酰基二苯酮(6),(6)在四乙酸铅 高氯酸 原甲酸三乙酯体系中重排、水解得酮基布洛芬.在此合成工艺中,格氏反应需无水无氧,反应条件苛刻,进行工业化生产很困难,且用四乙酸铅作重排试剂,使产品中铅的含量限度超过药典要求.为此,采用碘作重排试剂 2 ,使化合物(6)进行氧化性重排,经水解制得酮基布洛芬,总收率为28%.合成路线见图1.重排反应中,加入Lewis 酸可显著降低碘的用量.Fig 1 Route of synthes is在氮气保护下,化合物(2)与氰化亚铜反应,实验中将反应温度由文献 3 的155~170!升至170~180!,反应时间也由文献的6h 延长至8h,结果,化合物(3)的收率由文献的78 3%提高至90%以上.曾参照文献 3 的方法,将化合物(3)在氢氧化钾的乙醇水溶液中进行水解,水解时间由文第 8 卷 第4期1998年12月 总30期中国药物化学杂志Chinese Journal of M edicinal Chemi stry Vol 8 No 4p.302Dec 1998 Sum 30献的24h 延长至60h,未得到(4).后改用55%~60%的硫酸在101~103!下水解8h,顺利得到化合物(4),收率达89%.文献 3 用五氯化磷作酰氯化剂,在四氯化碳溶剂中,于40!反应30min 后,经减压蒸馏得化合物(5).实验改用氯化亚砜为酰氯化剂,二氯甲烷作溶剂,回流反应7h,回收溶剂后得(5),可不经减压蒸馏,直接与无水苯在三氯化铝催化下制得化合物(6),两步收率为70 3%.(文献 3 收率:61 2%).化合物(6)与碘、原甲酸三乙酯、催化量的Lew is 酸回流反应15h,再水解即得化合物(1),收率为60%.研究发现,此重排反应中,碘与(6)的摩尔比为2∀1方能发生重排.若在反应体系中加入催化量的Lew is 酸(如溴化亚铜、氧化亚铜等)可显著地使碘与(6)的摩尔比降至1∀1.1 实验部分熔点用毛细管法测定,温度未经校正.3 溴苯丙酮(2)的制备参考文献 1 方法制得.1 1 3 氰基苯丙酮(3)的制备在干燥的四口烧瓶中,加入化合物(2)60g(0 28mol)、氰化亚铜30 4g(0 34mol)、DMF 42 3mL,氮气保护下在170~175!搅拌8h.稍冷,倒入六水三氯化铁112 8g 和水169mL 的溶液中,加入浓盐酸28 2mL,于60~70!搅拌0 5h.冷却后,用甲苯提取(100m L #3),合并有机层,用5%碳酸钠溶液(100mL #2)、水(100mL #3)洗涤,无水硫酸钠干燥,减压回收溶剂,得暗红色油状物39 7g,收率:90%,直接用于下步反应.1 2 3 丙酰基苯甲酸(4)的制备于三口反应瓶中,加入21g (0 13mol)化合物(3)及55%~60%的硫酸65m L,于100~103!搅拌7h.稍冷,用10%氢氧化钠溶液调至pH 9,加活性炭2g,脱色0 5h,抽滤,滤液用4mol/L 盐酸调pH 1~2,有大量浅黄色固体析出.抽滤至干,冷水洗至中性,干燥得浅黄色固体(4)17 0g,收率:78%,mp 121~123!(文献 3 m p 119~124!).1 3 3 丙酰基二苯酮(6)的制备于干燥的三口反应瓶中,加化合物(4)17g(0 1mol)、氯化亚砜23 8g(0 2mol)、二氯甲烷180mL 、无水DMF 一滴,搅拌回流7h.减压回收二氯甲烷和氯化亚砜,得暗红色油状物(冷却后固化),将其溶于36mL 无水苯中备用.在另一干燥的反应瓶中,加入无水三氯化铝27 0g (0 2mol)、无水苯90mL,室温搅拌15min,滴加上述酰氯的苯溶液,滴毕,于75~80!搅拌回流2h.冷却,倒入270g 碎冰和27m L 浓盐酸中搅拌2h.分出有机层,水层用苯提取(50mL,35mL,35mL),合并有机层,用温水洗(100mL #2)至pH 6,无水硫酸钠干燥.常压回收尽苯,减压蒸馏,收集158~165!/400Pa 馏分,得浅黄色液体17g,收率:75%(文献 3 m p 28~33!).1 4 酮基布洛芬(1)的合成于干燥的三口反应瓶中加(6)11 9g(0 05mol)、碘25 4g(0 05mol)、原甲酸三乙酯35 8g 、氧化亚铜0 576g (0 004mol),搅拌回流15h.稍冷,加入10%连二亚硫酸钠60mL,搅拌10min,抽滤,滤液分出有机层,水层用三氯甲烷提取(20mL #3),合并有机层,用10%连二亚硫酸钠(50mL #1)、饱和氯化钠(50mL #2)、水(50mL #2)洗涤,无水硫酸钠干燥,减压回收溶剂,得棕褐色油状物.3033期户业丽等:碘重非法合成酮基布洛芬304中国药物化学杂志8卷将上述油状物与40mL甲醇及30mL30%氢氧化钠搅拌回流4h.补加水35m L、活性炭1g,回流脱色0 5h.抽滤,水洗,合并碱水层,用4mol/L盐酸调pH1~2,苯萃取(20mL# 3),合并有机层,水洗(50mL#3),无水硫酸钠干燥,回收苯,得黄色油状物.上述油状物与6mol/L盐酸30m L搅拌回流4h.冷却,分出有机层,水层用苯提取(20mL#2),合并有机层,加8%氢氧化钠调pH8,搅拌15m in,分出碱水层,有机层用8%氢氧化钠洗(20mL#2),合并碱水层,加活性炭(0 5g)脱色0 5h.抽滤,水洗,滤液用冰醋酸调pH1~2,有油状物析出.静置过夜,倾出水层,用丙酮溶解油状物,无水硫酸钠干燥,减压回收溶剂,得黄棕色油状物,放置固化,用苯 石油醚重结晶,得白色晶体(1)7 6g,收率:60%,m p 94~96!.1H NMR(D2O) :1 56(3H,d),3 78(1H,q),4 65(1H,s),7 2~7 8(9H,m).参 考 文 献1 陈芬儿,张文文.酮基布洛芬的合成.中国医药工业杂志,1991,22(8):344~3452 Huggins SD,Thomas CB.Conversi on of aromatic ketones into arylpropionic acids.J Chem Soc,Perkin T rans,I1983,(7):1483~14883 Zupancis Boris,Jenko Branko.The preparation of2 (3 benzoylph enyl)propionic acid.Ger Off.2913770.11,Oct.1979Synthesis of Ketoprofen via RearrangementUsing Iodine as the CatalystH u Yeli,Ling Xiuhong(Research Centre of Sy nthetic Dr ugs,Wuhan I nstitute ofChemical T echnology,Wuhan430074)Chen Fener(Dep ar tment of Chemistry,Fudan University,Shanghai200433)Abstract An efficient preparation of ketoprofen w as described.Propiophenone was used as the material via bromination,cyanation,hydrolysis,the Friedel Crafts∃reaction to give3 propionyl benzophenone,which w as converted to ketoprofen by rearrangement w ith iodine.The overall yield w as28%from propiophenone.Key words ketoprofen;rearrangement;synthesis。

收稿日期:2002-10-31作者简介:余红霞(1968-),女(汉族),湖北武汉人,讲师,硕士,主要从事药物及中间体的合成研究工作,Tel :(027)88025421,E 2mail :tree -2001-11@1631com 。
文章编号:1005-0108(2003)02-0097-02酮基布洛芬的合成方法改进余红霞1,郭峰2,陈芬儿3(11武汉化工学院制药系,湖北武汉430073;21中国药科大学药学院,江苏南京210009;31复旦大学化学系,上海200433)摘 要:目的合成酮基布洛芬,提高产品纯度。
方法以32苄基苯乙酮为原料,通过Darzens 缩合,氧化合成目标物。
结果与结论以32苄基苯乙酮计算,总收率约64%,产品纯度为9918%。
并通过红外吸收光谱(IR )、质谱(MS )确证了目标物的结构。
该合成工艺简单,产品质量好。
关键词:药物化学;工艺改进;Darzens 缩合;酮基布洛芬中图分类号:R914 文献标识码:A 酮基布洛芬是优良的非甾体消炎药,具有抗炎作用强、副作用小等特点,临床上用于治疗慢性类风湿关节炎、变形性关节炎、外伤和手术后疼痛。
自1973年上市以来,人们对其进行了大量研究,其合成方法很多,其中以Darzens 缩合法引人瞩目。
我国西南合成药厂首次采用此法实现了酮基布洛芬的生产,但生产实践表明,32苄基苯乙酮在进行Darzens 缩合过程中,除获得32苄基2α2甲基苯乙醛(3)外,尚有约10%~12%的反应物32苄基苯乙酮,难以用常规减压蒸馏的方法除去,当用高锰酸钾氧化3时,不可避免地使32苄基苯乙酮氧化成32苯甲酰基苯甲酸,该副产物与酮基布洛芬难以通过重结晶的方法加以分离,致使酮基布洛芬的质量不能达到英国药典1993年1995增补版标准。
为此在用Darzens 缩合反应来制备32苄基2α2甲基苯乙醛时,本文作者采用在磷酸二氢钠和过氧化氢体系中,用亚氯酸钠选择性将3氧化为4,而该物质为固体,从而除去32苄基苯乙酮,再氧化制得较纯的目标物1。

布洛芬剂型研究进展作者:李晓洁王崇静梁月琴王珩李仲昆来源:《中国实用医药》2014年第18期布洛芬的化学名为2-(4-异丁基苯基)丙酸,属丙酸衍生物,是一个已使用多年的老药。
其临床应用主要是解热,镇痛,抗炎。
但其长期口服可产生一些不良反应。
周晓梅等[1]收集了2000~2012年国内医药期刊报道的有关布洛芬的不良反应。
据统计,布洛芬致胃肠道不良反应居多;在治疗类风湿性关节炎、关节疼痛时引起不良反应的较多;老年人使用布洛芬出现不良反应的情况较多;在使用时间上,超时使用致不良反应的达23.53%;连续用药出现不良反应的占到52.94%。
针对这些不良反应,人们一直致力于其制剂和技术方面的研究,本文拟就布洛芬的制剂及剂型的研究进展作一综述。
1 ;片剂布洛芬原料药为白色结晶粉末,因其熔点低,与辅料混合后极易产生低共熔现象。
陈晓刚[2]以HPMC为粘合剂制粒,胶态SiO2和MS 混合物外配以解决上冲粘冲现象,外配滑石粉因重下沉以解决下冲粘冲情况,同时处方中加入了聚山梨酯80可确保布洛芬片溶出度达到国外药典的高标准要求。
2 ;乳膏布洛芬局部给药2~4 h,血药浓度达到峰值,在皮下脂肪、肌肉和关节滑液中的药物浓度远远超过血药浓度有利于局部炎症和疼痛的治疗。
符棘玉等[3]研究了氮酮对布洛芬乳膏体外透皮吸收的影响,发现当氮酮的浓度为3%时,布洛芬乳膏的透皮吸收率最高,可以选择氮酮作为布洛芬乳膏的促透剂。
3 ;滴眼液布洛芬难溶于水,因此很难配制成溶液剂型。
韩华等[4]利用布洛芬与聚乙二醇-20000形成低聚共熔混合物的方法,制备了0.2%的布洛芬滴眼液。
对168 例患各种类型结膜炎及内眼手术后患者的临床总有效率为97.0%,未发现明显的毒副作用。
4 ;巴布剂布洛芬的适合局部给药,国外已经成功将布洛芬开发成多种透皮制剂。
王霞等[5]将布洛芬制成药量为5%的巴布剂,所得布洛芬巴布剂含量稳定,渗透速率小于释放速率。


布洛芬新工艺研究引言布洛芬是一种具有解热、镇痛和抗炎作用的药物,被广泛应用于临床医学中。
随着科技的不断进步,对布洛芬生产工艺的研究也在不断深入。
传统的布洛芬生产工艺存在着一些不足之处,如生产效率低下、能耗高等。
因此,研究一种高效、环保的布洛芬新工艺具有重要意义。
本文将介绍布洛芬新工艺研究的现状、方法、结果与展望。
研究现状布洛芬新工艺研究的发展迅速,国内外研究者不断探索新的生产方法。
在国内外学者的共同努力下,一些新型的布洛芬生产工艺逐渐被开发出来。
例如,有学者采用生物酶法合成布洛芬,这种方法具有高效、环保等优点,但生物酶的来源和稳定性仍是亟待解决的问题。
另外,还有学者尝试采用有机合成的方法生产布洛芬,但这种方法生产过程较长,成本较高。
因此,针对现有布洛芬生产工艺存在的问题,本文将重点研究一种新型的布洛芬生产工艺。
研究方法本研究将采用文献调研和实验研究相结合的方法,首先通过文献调研了解布洛芬生产工艺的研究现状和进展,然后通过实验研究探索新型布洛芬生产工艺的可行性。
具体实验方案如下:1、材料:准备布洛芬原料、催化剂、溶剂等实验材料。
2、方法:采用液相催化氢化法合成布洛芬,通过调节反应温度、压力、催化剂用量等参数,探究最佳反应条件。
3、实验设计:进行不同条件下催化剂的活性评价实验,采用正交试验设计方法优选最佳反应条件。
4、数据处理:对实验数据进行整理和分析,采用表格和图表记录数据。
研究结果通过实验研究,我们发现新型布洛芬生产工艺具有以下优点:1、反应条件温和,催化剂活性高,可有效提高布洛芬的产率;2、反应过程易于控制,产品质量稳定,可实现工业化生产;3、催化剂可循环使用,减少了对环境的影响。
然而,新型布洛芬生产工艺也存在一些不足之处:1、催化剂成本较高,可能会增加生产成本;2、工艺流程仍较长,需要进一步优化简化。
结论与展望通过对布洛芬新工艺的研究,我们发现新型生产工艺具有较高的生产效率和环保性能。
虽然新型生产工艺仍存在一些不足之处,但可以通过进一步优化催化剂、简化工艺流程等措施加以改进。

酮基布洛芬生产工艺研究与改进措施分析【摘要】酮基布洛芬是一种非甾体消炎、镇痛药物,为了提高药物生产的安全性和有效性,必须对原有的生产工艺进行优化和改进。
下面本文就对酮基布洛芬生产工艺进行研究,并对其优化和改进措施进行了分析。
【关键词】酮基布洛芬生产工艺改进措施酮基布洛芬是一种非甾体类药物,就有消炎、镇痛以及解热的功效,并且具有高效低毒、口服吸收快以及消除快的优点。
为能够提高药物的安全性以及有效性,就需要对之前的生产工艺不断进行优化和改进。
1 原有的生产工艺改进之前的工艺流程如图一所示。
其技术指标为:甲基化反应收率为50%~55%,其中间产物即3-氰乙基苯甲酸甲酯的纯度为93%~94.5%;酰化-傅克反应其收率为75%~80%,这一反应的中间产物,也就是3-苯甲酰基-α-甲基苯乙腈的纯度要>93.4%;脂水解反应的收率要>94%,其中间产物,也就是3-氰乙基苯甲酸的纯度为95%~96%;氰水解反应的收率为83%~87%,其产物酮洛芬的纯度即为98.39%~99.4%(如图1所示)。
2 存在的问题采用以上生产工艺生产酮基布洛芬,如果采用的是间氰甲基苯甲酸甲酯作为原料进行生产,在经过了一系列的反应之后,其酮基布洛芬的收率仅为32%~39%。
经过分析,出现这一问题的原因主要是在甲基化反应以及酰化-傅克反应上,主要是因为这两个环节的反应收率比较低引起的。
其中在甲基化反应中,其单甲基化试剂采用的是硫酸二甲酯,那么就会造成双甲基化产物的出现,从而对单甲基化的选择率产生影响;另外在酰化-傅克反应环节的问题主要是因为酰氯物中的SOCl2没有蒸馏完毕,因而造成了二苯亚砜副产物的产生。
所以对于原有生产工艺的改进,改进重点应该放在对这两个反应环节上。
3 改进措施(1)对于甲基化反应的改进和优化。
如果是在强碱和相转移催化剂共同存在下,采用(CH3)2SO2作为甲基化的试剂,然后在芳基乙腈的侧链-CH2CN 上把甲基和α-单甲基化的时候,会有双甲基化副产物的出现,并且很难和双甲基化进行纯化分离。

合剂制备的西尼地平片成型较好%设计如下处方进行试验%原辅料处方&-’处方一处方二处方三处方四西尼地平7(57(57(57(5乳糖63(56=(568(568(5羟丙甲纤维素7666十二烷基硫酸钠7767吐温B>77776硬脂酸镁7(87(87(87(8!!按以上四个处方分别称量$制粒$压片!以片面外观$硬度$崩解时间及溶出度作为主要考察指标!结果处方一$二$三的片面外观及硬度均符合要求!处方四片面外观黄色!硬度不能达到要求%处方一的片子=7E a1时未能完全崩解!处方二$三$四的崩解均较好!约@E a1左右崩解完全%以7( =?十二烷基硫酸钠溶液作溶出介质时!处方二$三在=7E a1时均能溶出D5?以上%考虑到处方三比处方二工艺简单!更易于制粒$压片!因此选用处方二作为西尼地平片的处方!继续进行考察%经溶出度试验!按处方二制备的西尼地平片溶出较快!在7(5?十二烷基硫酸钠溶液介质中!桨法!转速)57]*E a1B6!35E a1内能溶出D5?以上%经稳定性试验!本品对光$湿$热的稳定性较好%"!规格与处方8C6规格!5E-+片%8C8处方!每6777片量%原辅料用量西尼地平5-乳糖6=5-羟丙甲纤维素67-硬脂酸镁8-65?聚乙烯吡咯烷酮溶液约6>E F 8C=处方中各组份的作用西尼地平!!!!!!!主药乳糖!!!!!!!!!填充剂羟丙甲纤维素!!!!!崩解剂硬脂酸镁!!!!!!!润滑剂聚乙烯吡咯烷酮!!!!粘合剂#!西尼地平片的制备工艺=C6先将西尼地平$乳糖$羟丙甲纤维素分别粉碎!过677目筛!备用%=C8按处方称取西尼地平$乳糖$羟丙甲纤维素!以等量递加法混合均匀%加入粘合剂65?聚乙烯吡咯烷酮溶液制软材!87目筛制粒!颗粒摊成薄层97f下烘干!87目筛整粒!加入硬脂酸镁!混匀%=C=取样化验颗粒主药含量!根据所测颗粒含量!计算片重!压片%=C3取样全检!合格后包装得成品%$!结论综合以上研究!对西尼地平片剂的不同处方进行了筛选!并对各项指标进行了考察!确定了西尼地平片剂最佳处方和工艺%参考文献/60罗明生!高天惠C药剂辅料大全C成都)四川科学技术出版社!6@@=C/80南京药学院药学教研组C药剂学C北京)人民卫生出版社!6@>5C /=0奚念朱C药剂学!北京)人民卫生出版社!8777C布洛芬产品中重排工艺改进舒瑞友$山东新华制药股份有限公司研究院!淄博!855775%摘要&改进布洛芬生产中的重排反应工艺!方法!新工艺使用液体布洛芬锌替代氧化锌为重排催化剂#并且通过正交试验对工艺进行优化设计!结果及结论!新较以往的反应#有反应温度降低"时间缩短"易控制"粗品颜色浅#收率达到>>?等优点#易达到工业化生产的要求关键词&布洛芬!工艺改进!布洛芬锌中图分类号&67-&,1&!文献标识码&2!文章编号&%&’()’’*+"(,,&#,-),(-(),*F7C6;V47495;<F H D C6;<4956:9?<;6789A>6:<5<Pi K c a:^.c$<$,1+.1-V a1$c,4$,]E,0\c%a0,F H a E a%\+#.E&,1^#U a‘.#855775%)J/(&)3(!’J K*3(F L*!M.a E&]./\%$\%],1)*.]E a1-0],*%a1!‘c&].*\1&].+c0%a.1C M*(1’-/!H a e c a+!‘c&].*\1U a10’,))c‘)%a%c%\+U a10.b a+\’.][,)0,%,F^)%!,1+0,]]a\+.1%$\.]%$.-.1,F\b&\]a E\1%%..&%a E a k\+%$\%\0$1a e c\+\)a-1(&*U /N2(/:9B3’O32N/F’O!#.E&,]\+%.%$\.F+E.%$\+)!%$\1\’’,^0,1+\0]\,)\%$\]\,0%a.1M\E&\],%c]\,1+]\,0%a.1 万方数据%a E\!a E&]./\%$\e c F a%^.*&].+c0%)(M$\],%a.0,1,0$a\/\%.>>?!,1+‘\)c a%*.]a1+c)%]a,F a k,%a.1( P*Q R’&-/!!‘c&].*\1(#],*%a E&]./\E\1%(!‘c&].*\1U a10!!布洛芬为非甾体类解热镇痛药!疗效确切!其生产工艺和萘普生相似!萘普生工艺中的重排反应使用的是萘普生锌作催化剂!我们参照此工艺制备出了布洛芬锌!应用于布洛芬重排反应中!实现了工艺优化%!!实验部分6(6布洛芬锌的制备!在577E F的反应瓶中!装好搅拌器和温度计!加入693(@9-布洛芬!搅拌升温至熔融透明后!加入氧化锌65(5-!搅拌升温@5f!675f!反应35E a1!加入877E F甲苯搅拌下升温并溶解至透明!回流带水8$!测水分!水分含量7(8?以下为合格!不合格时要继续带水直到合格%6(8合成路线6(=缩酮合成布洛芬过程!在857E F反应瓶上装好搅拌及回流分水装置!加入配量的缩酮=6(75-&缩酮溶于=7E F甲苯液’!再加入甲苯37E F$二甲苯677E F!升温至6=7f!滴加布洛芬锌3(D9-&溶于67E F甲苯中’!5E a1加完!通过加热和从分水器中放掉一部分溶媒使反应温度稳定在6=7f!637f!反应6(5!8$后停止反应!降温至97!D7f后溶液滴加57?W,Y P&67-!67E F水’约5E a1加完%继续升温至>5!675f水解反应8(5$降温至D7!D5f加入晶种&布洛芬钠’有结晶析出后!停止搅拌自然降温至室温!然后用冰水浴降温至67f左右!过滤用甲苯洗涤=!3次!每次57E F!至洗液无色为止%抽干后晾干!送样分析Y P B及钠盐含量!含量在>7!@7?之间!钠盐重约3=!39-!为白色片状结晶(将钠盐溶于877E F97f热水中!用盐酸调节&P至D(5!>!这时析出大量的氢氧化锌沉淀!滤液降温到常温时!慢慢滴加盐酸到&P小于8!停止滴加盐酸继续搅拌8$!过滤得湿品布洛芬!97f干燥得布洛芬%6(3对重排工艺通过H&=3’正交实验设计表选择最佳工艺控制参数)表!正交实验因素位级表位级因素2)催化剂用量&-*-B6’A)反应温度&f’#)反应时间&$’")缩酮含水量&-*-B6’16(7!8(7687!6=77!7(57!7(6 #7(5!6(76=7!6376(7!6(57(6B7(5 28(7!3(7637!657=(7!3(77(5!6(5!!表"位级因素2)催化剂用量&-*-B6’A)反应温度&f’#)反应时间&$’"(缩酮含水量&-*-B6’收率?666=8>3(D>88666>3(98==68=>D(3>36886@6(67588==>5(379=868>>(6>D6=6=>=(63>8=88>8(@9@===6>8(D9 Z685@(78859(>>855(@385>(3>Z8858(@>893(9>896(53855(D8Z=85>(3883>(>9858(@3859(78Z6+=>9(=3>5(9=>5(=6>9(69Z8+=>3(==>>(8=>D(6>>5(83Z=+=>9(63>8(>@>3(=6>5(=3K8(765(858(>D7(@8"!结果通过正交实验得出结论)以上四个因素的最佳组合是26$A8$#8$"6(从极差分析来看!提高收率的因素主次依次为A’#’2’"!见图6!以原工艺和最佳组合工艺各做了六批收率!结果见图8)图6!正交实验因素趋势图图8!原工艺和优化工艺图#!结论讨论=C6优化组合工艺即!反应温度6=7!637f$反应时间6! 6(5$$催化剂用量占投料量的6?!8?$原料缩酮水分6m 以下!缩酮到布洛芬收率从原工艺的>8(6?提高到>>( =9?!产品质量符合i<4标准%=C8布洛芬锌催化剂参入的重排反应是均相液体回流反应!反应温度低!反应时间短!因而操作简单稳定!粗品布洛芬颜色浅!为类白色(原工艺氧化锌参入的重排反应为固液非均相反应!反应温度为657!697f!反应时间多达5!9$!操作不稳定!粗品布洛芬颜色为黄色!质量差% 万方数据=C=缩酮中含水量超过6m!很容易产生逆反应!生成氯酮使收率降低%分离纯化野马追总黄酮精制工艺中大孔树脂的筛选方法张丽梅$山东省医药工业研究所!济南!857677%摘要&目的!筛选野马追总黄酮的精制工艺中分离纯化效果最好的大孔吸附树脂!方法!以总黄酮含量为评价指标#考察静态吸附"解吸附"动态洗脱性能等参数#对"676型""876型""=76型""376型等大孔吸附树脂吸附分离纯化野马追总黄酮进行评价!结果!"=76型大孔吸附树脂静态饱和吸附量为6>(3>E-)-B6’在97?">7?乙醇中静态解吸附率为>=(99?和>5(=9?#乙醇洗脱时动态解吸附率为@=(D7?#综合性能较好!结论!"=76型大孔吸附树脂综合性能最好#适合野马追总黄酮的精制!关键词&大孔吸附树脂!分离纯化!筛选中图分类号&!(+-1(!文献标识码&2!文章编号&%&’()’’*+"(,,&#,-),(--),*/4S4>55=4H4?57:>6;C;6;D?64?895;?4C:6:54:9B C D68<I146H:*D C:5;688U P2W;H a:E\a$<$,1+.1-!1)%a%c%\.*4$,]E,0\c%a0,F!1+c)%]^#T a j1,1#857677%)J/(&)3(!’J K*3(F L*!<\F\0%%$\‘\)%%^&\).*E,0].&.].c)]\)a1%.&c]a*^P\]‘,X c&,%.]a a(M45=;B?!"676$"876$ "=76$"376%^&\).*E,0].&.].c)]\)a1’\]\c)\+%.)\&,],%\,1+&c]a*^%.%,F*F,/.1\)(M$\)%,%a0,+).]&%a.10,&,0a%^!%$\)%,%a0 \F c%a.1],%a.!%$\+^1,E a0,+).]&%a.10,&,0a%^’\]\0.E&,]\+(&*/N2(/!M$\)%,%a0,+).]&%a.10,&,0a%^.*"=76E,0].&].c)]\: %a.1’,)6>(3>E-*-B6!%$\\F c%a.1],%a.’,)>=(99?!>5(=9?!]\)&\0%a/\F^(M$\+^1,E a0,+).]&%a.10,&,0a%^’,)@=(D7?( 3’O32N/F’O!M$\"=76%^&\E,0].&].c)]\)a1)$.’\+‘\%%\]0.E&]\$\1)a/\,+).]&%a.1&].&\]%^!a%0,1‘\c)\+%.&c]a*^%$\ \b%],0%.*P\]‘,X c&,%.]a a(P*Q R’&-/!E,0].&.].c)]\)a1()\&,],%a.1,1+&c]a*a0,%a.1()0]\\1a1-!!大孔树脂是一类不带离子交换基团的多孔型交联聚合物!对化学物质的分离作用主要由其吸附性产生!将大孔树脂应用于中草药的分离纯化!在我国始于D7年代!近年来广泛应用于天然产物的分离%其中对单味中药提取物中苷类成分!如黄酮苷等的分离纯化效果较好%本试验对野马追中黄酮类化合物进行分离纯化%!!仪器与试药"iB937紫外分光光度计(芦丁对照品&中国药品生物制品检定所’("676型$"876型$"=76型$"376型$2AB>型$U M#B6型大孔树脂&天津树脂厂’(甲醇$无水乙醇等试剂均为分析纯%"!试验方法和结果8(6大孔树脂处理方法!取大孔树脂!加入@5?乙醇浸渍&高于树脂层670E的体积’3$!装柱!用@5?乙醇淋洗至清&6E F流出液加=E F蒸馏水不浑浊’!再用蒸馏水反复洗涤至无醇味%将洗涤干净的大孔树脂抽干!备用%8(8测定波长的选择!分别称取野马追乙醇提取液$芦丁对照品溶液!以空白为对照!在377!9771E波长范围内扫描%结果显示)野马追乙醇提取液和芦丁对照品的最大吸收波长分别在3@71E和3>D1E!故选择在3>D1E处测定样品吸收度% 8(=标准曲线的制备!标准溶液制备)分别精密量取芦丁对照品贮备液7(5$6(7$8(7$=(7$3(7$5(7$9(7E F!置于85E F容量瓶中!加蒸馏水至67E F!加5?W,W Y86E F!放置9E a1!再加67?2F&W Y=’=6E F!放置9E a1!加6E.F*H B6W,Y P 67E F!加蒸馏水定容至刻度%以所加试剂为空白对照!65E a1后!在3>D1E波长处测定吸收度%以芦丁含量为横坐标!吸收度为纵坐标!计算得回归方程为)J_7(336>K B7(778D& C_7(@@@>’!芦丁线性范围为7(67=95E-!6(83=>E-%8(3不同吸附树脂对野马追总黄酮静态吸附实验!精密称取已处理好的"676型$"876型$"=76型$"376型$2AB>型$ U M#B6型大孔树脂各6-!分别置85E F烧瓶中!各加野马追乙醇提取液67E F!每隔5E a1振摇67)!持续8$%静置83$!使其达到饱和吸附!吸取7(5E F的上层液于85E F容量瓶中!制备供试品溶液%在3>D1E波长处测定各溶液的吸收度!根据标准曲线计算总黄酮含量!并按下式计算树脂饱和吸附量)饱和吸附量&比吸附率’_/&初始浓度B吸附后浓度’J吸附液体积0+树脂量&E-*-B6’溶液中总黄酮测定结果见表6% 万方数据。

生物催化法制备手性酮基布洛芬的研究进展胡琦蔚;张俊伟;王远山【摘要】酮基布洛芬是一种重要的消炎镇痛药,广泛应用于风湿、类风湿性关节炎、脊髓炎和痛风等疾病的治疗,成为处方量最大的药物之一.市售的酮基布洛芬以外消旋体为主,(S)-酮基布洛芬为其活性对映体,(R)-酮基布洛芬的存在会加重肝脏的代谢负担.目前手性酮基布洛芬主要通过化学合成法生产,与传统的化学法相比,生物催化法具有反应条件温和、对环境友好等优点,成为近年来研究的热点.生物催化法主要利用脂肪酶、酯酶和腈水合酶/酰胺酶双酶体系等催化制备手性酮基布洛芬.同时对生物催化法制备手性酮基布洛芬的研究进展进行了综述.【期刊名称】《发酵科技通讯》【年(卷),期】2017(046)003【总页数】5页(P153-157)【关键词】生物催化;酮基布洛芬;脂肪酶;酯酶【作者】胡琦蔚;张俊伟;王远山【作者单位】浙江工业大学生物工程学院,浙江杭州310014;杭州安诺过滤器材有限公司,浙江杭州311404;浙江工业大学生物工程学院,浙江杭州310014【正文语种】中文【中图分类】Q814酮基布洛芬(ketoprofen,3-苯甲酰基-α-甲基苯乙酸)是一种2-芳基丙酸类强效非甾体抗炎药.最早由法国化学家Rhone Poulenc于1967年合成,并于1973年引入法国和美国作为抗炎药使用[1],对风湿、类风湿性关节炎、脊髓炎和痛风等疾病有良好的效果,且消炎作用强于布洛芬,不良反应轻于布洛芬.在同等剂量下,其消炎镇痛作用是阿司匹林的150倍,解热作用是消炎痛的4倍、阿司匹林的100倍.由于酮基布洛芬具备药效高、半衰期较短、代谢简单和不良反应少而轻等优点,已广泛应用于治疗各种类型的疼痛、炎症症状、感冒及手术后的消炎止痛.由于风湿、类风湿和痛风等是我国多发性疾病,近年来酮基布洛芬等非甾体抗炎药发展迅速,市场需求量巨大.而酮基布洛芬具有“R”和“S”两种对映体,并有着明显不同的药理活性,(S)-(+)-酮基布洛芬(S-ketoprofen)是(R,S)-酮基布洛芬的活性对映体,其消炎镇痛等作用是外消旋体的两倍[2],而(R)-酮基布洛芬抗炎镇痛作用极弱,常作牙膏添加剂以预防和治疗由牙周炎导致的骨质疏松[3],也可用于缓解触觉性异常疼痛.目前,市售的以外消旋体为主,疗效较(S)-(+)-酮基布洛芬弱且肝脏代谢负担大.因此,开发低成本的单一对映体的(S)-(+)-酮基洛芬制备工艺具有重要意义.1.1 结晶法1.1.1 非对映体结晶法非对映体结晶法是利用拆分剂(如光学纯的酸)拆分外消旋体得到非对映体盐,再通过选择性结晶从而得到光学纯的对映体.Yoneyoshi等[4]将外消旋酮洛芬和酮洛芬胺加热溶解在甲醇中,冷却后静置过夜得到结晶,过滤后用1%的盐酸将结晶溶解,用甲苯萃取两次后减压蒸馏得到(S)-(+)-酮基布洛芬,产率为43%,e.e.值为79.1%.Lukas等[5]则用异丙醇溶解外消旋酮洛芬,加入苯乙胺作为手性试剂,搅拌后冷却结晶,用0 ℃的异丙醇洗涤结晶两次得到(S)-(+)-酮基布洛芬,产率为40%,文中未提及产物的e.e.值.1.1.2 优先结晶法优先结晶是在饱和或过饱和的外消旋体溶液中加入其中一种对映异构体的晶体作为晶种,造成不对称环境,诱导结晶按非平衡的过程进行,从而达到拆分的效果.Van Eikeren等[6]通过加热搅拌使外消旋的酮洛芬溶解在乙腈-水混合物中,以(1R,2S)-cis-1-aminoindan-2-ol作为拆分剂,加入(S)-ketoprofen-(1R,2S)-cis-1-aminoindan-2-ol作为晶种,得到的结晶用乙腈洗涤两次后真空干燥,最终获得66.4 g (S)-(+)-酮基布洛芬,e.e.值达到97.2%.Manimaran等[7]用甲醇稀释外消旋酮洛芬的乙酸乙酯溶液,冷却后加入(S)-ketoprofen-cinchonidine晶体,得到的结晶在真空条件下过滤,并用乙酸乙酯和乙醚洗涤三次,真空干燥后得到成品,(S)-(+)-酮基布洛芬的产率为31%,e.e.值为97%.由于利用结晶法制备(S)-(+)-酮基布洛芬的过程中均涉及重结晶这一步骤,耗时且增加了成本、降低了产率.另外,结晶中所用到的基质昂贵且不稳定,难以循环利用[8],制备过程中对温度和压力的要求也较高,尤其是结晶时,往往需要低温保证结晶的顺利进行,由于这些缺陷的存在,结晶法并未被广泛应用于工业生产中. 1.2 不对称合成法1.2.1 Sharpless环氧化法Hamon等[9]通过6步反应,由烯丙醇通过Sharpless环氧化制备得到(2S,3S)环氧化合物,再加入手性位移试剂,经过对映体选择性氢解后生成二醇,在RuO4/NaIO4的催化下,最终得到(S)-(+)-酮基布洛芬,e.e.值达到98%.Sharpless环氧化法制备(S)-(+)-酮基布洛芬的途径为[9]1.2.2 α-芳基丙烯酸氢化法具有前手性的不饱和羧酸经手性BLNAP-Ru的二羧酸酯络合物氢化后可得到光学活性的饱和羧酸,该方法可用于(S)-(+)-酮基布洛芬的制备.Laue等[10]以(S)-(+)-RuBLNAP(OAc)2为催化剂在高压下氢化,最终得到的产物e.e.值达到80%.利用手性络合物制备(S)-(+)-酮基布洛芬的途径[10]为目前,市售(S)-(+)-酮基布洛芬主要通过传统的化学法生产,这种方法的产率和对映体选择性较为理想,但是均需要用到传统的化学催化剂以促使反应快速进行,而大部分催化剂毒性较高、易燃且会造成环境污染,还有可能引入有害的副产物.另外,手性催化剂的制备过程较为困难,有时其本身的制备反应也需要昂贵催化剂的参与.酶法制备(S)-(+)-酮基布洛芬属于动力学拆分的范畴,由于其具有反应条件温和、较好的底物特异性和对映体选择性以及对环境友好等优点[11-13],受到了广泛的重视.生物催化法常以外消旋酮基布洛芬或其衍生物为底物,通过酶拆分得到(S)-(+)-酮基布洛芬.2.1 酯酶和脂肪酶酯酶和脂肪酶是(S)-(+)-酮基布洛芬制备中最为常用的酶.根据催化原理的不同,可以大致分为对映体选择性酯化和对映体选择性水解两类[14].2.1.1 对映体选择性酯化在对映体选择性酯化制备(S)-(+)-酮基布洛芬的过程中,通常以外消旋的酮洛芬和醇类作为底物,在具有选择性的酯酶或脂肪酶的催化下,优先将(R)-(-)-酮基布洛芬酯化为(R)-酮基布洛芬酯,而保留(S)-(+)-酮基布洛芬,再通过中和或加入饱和NaHCO3将剩余的酮洛芬与酮洛芬酯分离,以获得较纯的(S)-(+)-酮基布洛芬.通过对映体选择性酯化法制备(S)-(+)-酮基布洛芬的途径为Park等[15]利用Candida antarctica脂肪酶Novozym 435酯化外消旋的酮洛芬,发现以甲醇作为烷基供体时转化率和对映体选择性均较佳,转化9 h后,转化率达到59%,e.e.值为75%,E为6.Ong等[16]利用南极假丝酵母脂肪酶(C. antarctica lipase B,CALB,Novozym 525)拆分外消旋酮洛芬,比较了游离酶和固定化酶的催化特性,发现固定化酶在催化效率、热稳定性和重复利用等方面均要高于游离酶.以丁醇为烷基供体,在最佳条件下利用固定化酶反应24 h后,(R)-酮基布洛芬的转化率达到73%,剩余底物中(S)-(+)-酮基布洛芬的对映体过量值为87.8%.Candida cylindrecca所产脂肪酶(CCL)是用于选择性水解外消旋酮基布洛芬酯衍生物最有效的酶之一,以该酶为催化剂的报道较多,但其拆分反应需要大量手性环糊精参与.也有学者利用其他来源的酶进行对映体选择性酯化反应,如Li等[17]利用来源于Burkholderia cepacia的固定化脂肪酶G63,在37 ℃下反应22.5 h后得到(S)-(+)-酮基布洛芬,E为10.01.2.1.2 对映体选择性水解对映体选择性水解以外消旋的酮洛芬酯为底物,通过脂肪酶或酯酶优先水解其中一种对映体生成(S)-(+)-酮洛芬和醇.Kim等[18]从Archaeoglobus fulgidus DSM 4304基因组中克隆出一个耐高温的酯酶,结合易错PCR和定点饱和突变对该酶进行改造,得到一个双突变体V13G/L200R,在70 ℃下反应1 h,可以将5 mmol/L外消旋的酮基布洛芬乙酯转化为(S)-(+)-酮基布洛芬,e.e.值为(89.2±0.2)%,E为19.5±0.5.Kim等[19]从Candida rugosa中得到的脂肪酶可以将100 mmol/L酮洛芬乙酯转化为(S)-(+)-酮基布洛芬,e.e.值达99%,转化率为49%,但所需时间为3 d.Zhang等[20]利用固定化Trichosporon laibacchii酵母脂肪酶在含有Tween-80的水相中水解(R,S)-酮洛芬乙酯,45 ℃下反应33 h,转化率达到46.3%,产物的光学纯度达到94.4%.其余利用脂肪酶或酯酶制备(S)-(+)-酮基布洛芬的例子如表1所示.利用T. laibacchii脂肪酶水解(R,S)-酮洛芬乙酯生成(S)-(+)-酮基布洛芬[20]的途径为(R)-(-)-酮基布洛芬制备的相关研究较少.Gérard等[27]得到一个来源于酵母Yarrowia lipolytia的脂肪酶Lip2p,该酶可催化酮洛芬乙酯生成酮基布洛芬,但其对映体选择性不佳,对其进行定点饱和突变后获得一突变体V232F,可选择性催化底物生成(R)-(-)-酮基布洛芬,E≥300.Hu等[28]利用来源于Aspergillus terreus的脂肪酶催化酮洛芬乙烯酯,得到(R)-(-)-酮基布洛芬,转化率为(16.0±1.3)%,E为11.4,e.e.值为(82±2.7)%,后对其进行固定化,最大转化率、E和e.e.值分别提高到了45.9%,128.8和(96±0.1)%.2.2 腈水合酶/酰胺酶双酶体系目前也有利用腈水合酶和酰胺酶双酶体系制备光学纯酮洛芬的报道.Layh等[29]以酮洛芬腈为唯一氮原筛选得到的Rhodococcus equi K2a能将酮洛芬腈及酮洛芬酰胺转化为(S)-(+)-酮基布洛芬,e.e.值均为99%,转化率分别为27%和25%;Heinemann等[30]筛选得到的Agrobacterium sp. LK中含有的腈水合酶/酰胺酶双酶体系能够催化2-(3-苯甲酰苯基)丙腈转化成酮基布洛芬,e.e.值为91%;Salvo等[31]利用Agrobacterium radiobacter 30″60 (NCIMB 41108)静息细胞水解酮洛芬腈,(S)-(+)-酮基布洛芬的产率为45%,光学纯度为96%.利用腈水合酶/酰胺酶双酶体系制备(S)-(+)-酮基布洛芬[30]的途径为2.3 腈水解酶目前尚无利用腈水解酶制备(S)-(+)-酮基布洛芬的报道,但本课题组通过基因挖掘从NCBI数据库中挖掘得到一个腈水解酶Nit1,可选择性水解(R,S)-2-(3-苯甲酰苯基)丙腈生成(S)-(+)-酮基布洛芬,e.e.值为14.2%.对其进行分子改造后获得一个突变体W56G,该突变体的对映体选择性有了明显的提升.利用腈水解酶制备(S)-(+)-酮基布洛芬的途径为生物催化法具有条件温和,化学选择性、区域选择性及对映体选择性高等特点,且其对环境友好、耗能低,符合绿色发展的理念,建立酶法制备(S)-(+)-酮基布洛芬的新工艺,具有重要的意义.尽管在生物催化法制备(S)-(+)-酮基布洛芬方面进行了大量的研究,但由于酶拆分法存在着反应时间长、稳定性差等缺陷,该法还没有得到产业化应用.目前,已有较多通过对酶进行改造、固定化等手段从而改善了酶催化特性的例子,一定程度上突破了上述的限制,基因工程及酶工程的发展将极大推动生物催化制备(S)-(+)-酮基布洛芬技术的发展.【相关文献】[1] 石开云, 余清宝, 邹晓川. 酮洛芬合成方法研究进展[J]. 精细化工, 2015, 32(8):841-848.[2] HUTT A J, CALDWELL J. The importance of stereochemistry in the clinical pharmacokinetics of the 2-arylpropionic acid non-steroidal anti-inflammatory drugs[J]. Clinical pharmacokinetics, 1984, 9(4):371-373.[3] 赵运英, 刘瑞恩, 许丽娟, 等. 拆分获得(S)-酮基布洛芬脂肪酶基因在枯草芽孢杆菌中的克隆与表达[J]. 微生物学报, 2010, 50(5):634-640.[4] YONEYOSHI Y, KUDO J, NISHIOKA T. Optically active secondary amine compound, process for producing optically active secondary amine compound and process for producing optically active carboxylic acid by using said compound: US5510519[P]. 1994-03-29.[5] LUKAS H, SCHUSTER O, RAU G. Process to separate mixtures of enantiomeric arylpropionic acids: US 4983765 A[P]. 1991-01-08.[6] VAN E P, FRANCIS M X, LOPEZ J L. Process for resolving chiral acids with 1-aminoindan-2-ols: US 5677469 A[P]. 1997-10-14.[7] MANIMARAN T, POTTER A A. Resolution of ketoprofen: US5162576[P]. 1992-11-10.[8] BLASCHKE G, SCHULTE K E. Process for obtaining enantiomers of 2-arylpropionic acids: US 4973745[P]. 1990-11-27.[9] HAMON D P G, MASSY-WESTROPP R A, NEWTON J L. ChemInform abstract: enantioselective syntheses of 2-arylpropanoic acid non-steroidal antiinflammatory drugs and related compounds[J]. Cheminform, 1996, 27(11):12645-12660.[10] LAUE C, SCHRÖDER G, ARLT D. 2-Arylpropenic acids and their utilisation in the preparation of 5-ketoprofen: EP0529444[P]. 1997-04-16[11] WANG Y, LI Q, ZHANG Z, et al. Solvent effects on the enantioselectivity of the thermophilic lipase QLM in the resolution of (R,S)-2-octanol and (R,S)-2-pentanol[J]. Journal of molecular catalysis B enzymatic, 2009, 56(2/3):146-150.[12] 胡艾希, 董先明. 2-芳基丙酸类消炎药的酶催化拆分研究进展[J]. 中国医药工业杂志, 2001,32(6):284-288.[13] MCCOY M. Making drugs with little bugs[J]. Chemical & engineering news, 2010,79(21):37-43.[14] AI L O, KAMARUDDIN A H, BHATIA S. Current technologies for the production of (S)-ketoprofen: process perspective[J]. Process biochemistry, 2005, 40(11):3526-3535.[15] PARK H J, CHOI W J, HUH E C, et al. Production of optically active ketoprofen by direct enzymatic esterification[J]. Journal of bioscience & bioengineering, 1999, 87(4):545-547.[16] ONG A L, KAMARUDDIN A H, BHATIA S, et al. Enantioseparation of (R,S)-ketoprofenusing Candida antarcticalipase B in an enzymatic membrane reactor[J]. Journal of separation science, 2008, 31(31):2476-2485.[17] LI X, LIU T, XU L, et al. Resolution of racemic ketoprofen in organic solvents by lipase from Burkholderia cepacian G63[J]. Biotechnology and bioprocess engineering, 2012,17(6):1147-1155.[18] KIM J, KIM S, YOON S, et al. Improved enantioselectivity of thermostable esterase from Archaeoglobus fulgidus toward (S)-ketoprofen ethyl ester by directed evolution and characterization of mutant esterases[J]. Applied microbiology and biotechnology, 2015, 99(15):6293-6301.[19] KIM S H, KIM T K, SHIN G S, et al. Enantioselective hydrolysis of insoluble (R,S)-ketoprofen ethyl ester in dispersed aqueous reaction system induced by chiral cyclodextrin[J]. Biotechnology letters, 2004, 26(12):965-969.[20] ZHANG Y Y, LIU J H. Kinetic study of enantioselective hydrolysis of (R,S)-ketoprofen ethyl ester using immobilized T. laibacchii lipase[J]. Biochemical engineering journal, 2011, 54(1):40-46.[21] MIN G K, LEE E G, CHUNG B H. Improved enantioselectivity of Candida rugosa, lipase towards ketoprofen ethyl ester by a simple two-step treatment[J]. Process biochemistry, 2000, 35(9):977-982.[22] LONG Z D, XU J H, ZHAO L L, et al. Overexpression of Serratia marcescens lipase in Escherichia coli for efficient bioresolution of racemic ketoprofen[J]. Journal of molecular catalysis B enzymatic, 2007, 47(3):105-110.[23] SATHISHKUMAR M, JAYABALAN R, MUN S P, et al. Role of bicontinuous microemulsion in the rapid enzymatic hydrolysis of (R,S)-ketoprofen ethyl ester in a micro-reactor[J]. Bioresource technology, 2010, 101(20):7834-7840.[24] WANG Y H, YANG B, REN J, et al. Optimization of medium composition for the production of clavulanic acid by Streptomyces clavuligerus[J]. Process biochemistry, 2005, 40(3/4):1161-1166.[25] ZHANG W W, JIA J Q, WANG N, et al. Improved activity of lipase immobilized in microemulsion-based organogels for (R,S)-ketoprofen ester resolution: long-term stability and reusability[J]. Biotechnology reports, 2015, 7:1-8.[26] YOON S, KIM S, PARK S, et al. Improving the enantioselectivity of an esterase toward (S)-ketoprofen ethyl ester through protein engineering[J]. Journal of molecular catalysis B enzymatic, 2014, 100(4):25-31.[27] GÉRARD D, GUÉROULT M, CASAS-GODOY L, et al. Efficient resolution of profen ethyl ester racemates by engineered Yarrowia lipolytica Lip2p lipase[J]. Tetrahedron asymmetry, 2017, 28(3):433-441.[28] HU C, NA W, ZHANG W, et al. Immobilization of Aspergillus terreus lipase in self-assembled hollow nanospheres for enantioselective hydrolysis of ketoprofen vinyl ester[J]. Journal of biotechnology, 2015, 194:12-18.[29] LAYH N, KNACKMUSS H J, STOLZ A. Enantioselective hydrolysis of ketoprofen amide by Rhodococcus sp. C3II and Rhodococcus erythropolis MP 50[J]. Biotechnology letters, 1995, 17(2):187-192.[30] HEINEMANN U, KIZIAK C, ZIBEK S, et al. Conversion of aliphatic 2-acetoxynitriles by nitrile-hydrolysing bacteria[J]. Applied microbiology and biotechnology, 2003, 63(3):274-281.[31] SALVO G, BRANDT A, CECCHETELLI L. A micro-organism possessing enantioselective and regioselective nitrile hydratase/amidase activities: EP1291435[P]. 2003-03-12.(责任编辑:朱小惠)。

酮洛芬的工艺合成1产品简介1.1中英文名称,分子式,结构式名称:酮洛芬英文名:ketoprofe化学名:3-苯甲酰基-a-甲基苯乙酸分子式:C16H15O3结构式:OCOOHCH31.2 物化性质白色结晶性粉末;无臭或几乎无臭。
在甲醇中极易溶,在乙醇、丙酮或乙醚中易溶,在水中几乎不溶。
熔点约93—96℃。
为芳基烷酸类化合物。
1.3 用途常用的非甾体抗炎镇痛药,具有抗炎,解热,镇痛作用。
1.4 前景分析酮洛芬为一种强效非甾体抗炎药,是近年来应用于临床的高效解热,镇痛药物。
对风湿,类风湿性关节炎,脊髓炎,痛风等疾病有良好的效果。
随着国内经济的快速发展,酮洛芬药物生产技术不断提升,酮洛芬行业发展迅速。
与此有关的药物如:酮洛芬贴剂,酮洛芬缓释胶囊,酮洛芬分散片等药物得到广泛的生产应用。
国内企业为了获得更大的利润收益,在生产规模和产品质量上不断提高,与之相关的核心生产技术应用与研发成为业内企业关注的焦点。
技术工艺的优劣直接决定企业的市场竞争力,相应的酮洛芬合成的工艺流程不断地得到进一步的改进和提高。
2合成方法2.1第一种合成方法(1)合成原理以苯甲酸为起始原料,经溴化、Friedel-crafts反应、Grignard反应制得3乙酰基二苯酮,再经Darzens反应制得酮基布洛芬。
COOHCOOHBr1)pcl 5O CH 3OO COOHCH 3(2)合成所需要的原料苯甲酸,铁粉, 溴,五氯化磷,无水三氯化铝,镁粉,醇钠的乙醇溶液,氯乙酸乙酯乙腈,无水四氢呋喃(3)生产工艺,包括原料的用料比例,反应条件等。
第一步:3-溴苯甲酸的制备在干燥的四颈瓶中,加入73.2 g(0.6 mol)化合物苯甲酸、4g 还原铁粉,于100℃~150℃搅拌0.5h ,滴加18 mL(0.35 mol)溴,加毕搅拌1 h ,追加4g 铁粉和18 mL 溴,于150℃搅拌2h 后,再于260℃搅拌3h ,将产物溶于稀碱中,过滤后,用盐酸酸化使其析出晶体,重结晶,干燥得白色粉末固体第二步:3-溴二苯甲酮的制备在干燥的三颈瓶中,依次加入化合物3-溴苯甲酸55g(0.275mol)、五氯化磷59.8(0.333mol)及无水苯187.5mL,于80℃搅拌回流2 h ,用冰盐浴冷却至5℃以下,缓慢加入无水三氯化铝71.9g(0.95mol),温度控制在10℃以下,约1h 加完,在45~60℃搅拌回流5 h ,冷至室温后,将反应液倾入冰(500g )和浓盐酸(200mL )的混合液中,分出有机层,水层用苯提取(100mL ×2).合并有机层,依次用饱和碳酸氢钠溶液200mL 和水(200mL ×2)洗至中性,无水硫酸镁干燥,常压回收溶剂,得淡黄色固体70g 。
化学试剂,2006,28(5),316~317生产与提纯技术(R,S ) 酮基布洛芬的拆分研究刘成*1,刘志雄2,李翔2(1.广东光华化学厂有限公司广东省化学试剂工程技术研究开发中心,广东广州 510280;2.湖北省化学研究院,湖北武汉 430074)摘要:以N 辛基 D 葡糖胺为拆分剂,研究了(R,S) 酮基布洛芬的拆分及碱解,以及(R ) 酮基布洛芬的消旋;单轮拆分率达40%。
关键词:(R,S ) 酮基布洛芬;N 辛基 D 葡糖胺;拆分;消旋化中图分类号:O625 文献标识码:A 文章编号:0258 3283(2006)05 0316 02收稿日期:2005 09 20作者简介:刘成(1978 ),男,江西吉安人,硕士生,主要研究方向为精细化工。
(R,S ) 酮基布洛芬是非甾体抗炎药,有镇痛、消炎作用,用于治疗风湿性关节炎、骨性关节炎、脊椎炎以及软组织损伤,如腱炎和粘液囊炎。
其中的活性对映体(S ) 酮基布洛芬酮于1996年由意大利Menarini 公司开发并首次在西班牙上市,其消炎镇痛等作用是外消旋体的两倍,临床上只需外消旋体的一半用量即可达到相同的治疗效果。
获取(S ) 酮基布洛芬的方法主要有不对称合成、酶拆分、非对映体结晶拆分,其中不对称合成和酶拆分在我国现阶段尚未具备生产条件,而用手性胺的非对映体结晶拆分法具有操作简便、稳定等优点。
文献[1~5]使用辛可尼丁, 苯乙胺类, 苯丙胺等手性胺作拆分剂,但它们价格昂贵且来源不足。
N 辛基 D 葡糖胺是一种国内易得、价格较便宜的手性胺,已成功地用于萘普生[6,7]布洛芬[8]的工业拆分。
本文以N 辛基 D 葡糖胺为拆分剂,探讨了半量拆分法拆分(R,S ) 酮基布洛芬的方法。
CO CHCOOHCH 3(R ,S ) 酮基布洛芬1 实验部分1 1 主要仪器与试剂Varian Mercury VX 300型核磁共振仪;Impact 420型傅里叶红外光谱仪;Agilent 1100Series 高效液相色谱仪。
专利名称:含有酮基布洛芬的注射剂及其制备方法专利类型:发明专利
发明人:汪洪湖
申请号:CN200610044528.4
申请日:20060602
公开号:CN101077336A
公开日:
20071128
专利内容由知识产权出版社提供
摘要:本发明提供了一种含有酮基布洛芬的注射剂及其制备方法,其特征在于其中含有主药酮基布洛芬、碱性助溶剂和填充剂,酮基布洛芬与碱性助溶剂的摩尔配比为1∶1;酮基布洛芬与填充剂的重量比为1∶0-10。
该制剂克服传统口服酮基布洛芬制剂(如片剂、胶囊、分散片、口服混悬液等普通制剂)存在的溶解度小、吸收慢、生物利用度低、起效相对较慢的不足,以及对儿童、老年人和不能吞服固体制剂的患者带来不便;该制剂解决了产品溶解性及给药途径问题,所得的产品质量稳定、生物利用度高、起效迅速的优点。
申请人:汪洪湖
地址:233000 安徽省蚌埠市红旗二路西苑小区1号楼505号
国籍:CN
代理机构:安徽省蚌埠博源专利商标事务所
代理人:杨晋弘
更多信息请下载全文后查看。