gaussian高级注释
- 格式:pdf
- 大小:189.97 KB
- 文档页数:10

1“零点能”是指:量子在绝对温度的零点下仍会保持震动的能量,这个振动幅度会随着温度增加而加大。
“零点能”就是原子核旋转惯性能。
我们生活生产除核中子间斥力能外都是利用的核旋能,包括身体发热所需能量等,当然也包括燃烧、发光、发热和“磁”线圈产生的“磁力”。
零点能是对分子的电子能量的矫正,表明了在0K 温度下的分子的振动能量对电子能量的影响。
当比较OK 时的热动力学性质时,零点能需要被加到总能量中。
2密度泛函理论的基本原理是: 体系的基态能量是由电子的密度唯一确定的,其基本方程为Kohn-Sham方程, 它与HF方程在形式上完全一样, 只不过是用交换相关泛函代替HF的交换部分而已.在原理上是可以精确计算的,如果可以确定它的精确泛函的话。
但是, 由于其泛函没有一套系统的方法来逼近精确泛函,因此其必须从经验来确定泛函.这就是DFT近似的根源。
3 当体系变的松散时,ab initio 和DFT 基本上得不到最稳定的结构,你可以先用分子力学去优化一下。
或者你需要固定的原子是不是太大,如果可以作为环境处理的话,可以考虑QM/MM。
另外,初次优化基组不要用太大,逐步提高。
4如果你计算freq有个很小的虚频,可以用改变网格来消虚频。
默认的网格是75302,加入这个命令是指定99590网格。
5 L9999 出错就是说在默认的循环次数里未完成所要求的工作,无法写输出文件。
6一般地,优化所得驻点的性质(极小点还是过渡态)要靠频率来确定;而对过渡态,要确定反应路径(即到底是哪个反应的过渡态)必需要做IRC 了,不然靠不住的(往往用QST 找到的过渡态并不一定就是连接输入反应物和产物的过渡态)。
7在我们用QST2 或QST3 来优化过渡态时,需输入反应物和产物,实际上反应物和产物的输入顺序是没有关系的。
就是说,先输反应物后输产物和先输产物后输反应物得到的是同样的过渡态。
这也好理解,QST2 里对过渡态的初始猜测实际上是程序自动将输入的反应物和产物的各变量取个平均,所以输入顺序是没有关系的。

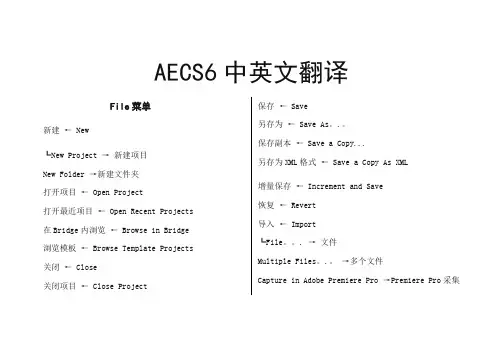
AECS6中英文翻译File菜单新建← New┗New Project →新建项目New Folder →新建文件夹打开项目← Open Project打开最近项目← Open Recent Projects 在Bridge内浏览← Browse in Bridge 浏览模板← Browse Template Projects 关闭← Close关闭项目← Close Project 保存← Save另存为← Save As。
.。
保存副本← Save a Copy...另存为XML格式← Save a Copy As XML增量保存← Increment and Save恢复← Revert导入← Import┗File。
. →文件Multiple Files。
.。
→多个文件Capture in Adobe Premiere Pro →Premiere Pro采集Adobe Clip Notes Comments。
. →Adobe 剪辑注释评论Adobe Premiere Pro Project…→Premiere Pro项目…Vanishing Point (vpe)…→ PS消失点(。
vpe)…Plac eholder.。
→输入占位符Solid... →实色导入最近素材← Import Recent Footage输出← ExportAdobe Dynamic Link Adobe ←动态链接查找← Find.。
.再次查找← Find Next添加素材到合成← Add Footage to Comp选定脚本建立合成← New Comp From Selection。
整理素材← Consolidate All Footage删除未用素材← Remove Unused Footage简化项目← Reduce Project 文件打包← Collect Files.。
. 浏览文件夹← Watch Folder。

收敛失败是很正常的事也是很头痛的事,在Gaussian98高级注释一文中提到以下几种做法:我经常用的是3/5/8/12,而且如果你是计算过渡金属,降低收敛标准你就得小心。
1. 在Guess关键字中使用Core,Huckel或Mix选项,试验不同的初始猜测。
2. 对开壳层体系,尝试收敛到同一分子的闭壳层离子,接下来用作开壳层计算的初始猜测。
添加电子可以给出更合理的虚轨道,但是作为普遍的经验规则,阳离子比阴离子更容易收敛。
选项Guess=Read定义初始猜测从Gaussian 计算生成的checkpoint文件中读取。
3. 另一个初始猜测方法是首先用小基组进行计算,由前一个波函得到用于大基组计算的初始猜测(Guess=Read自动进行)。
4. 尝试能级移动(SCF=Vshift)。
5. 如果接近SCF但未达到,收敛标准就会放松或者忽略收敛标准。
这通常用于不是在初始猜测而是在平衡结构收敛的几何优化。
SCF=Sleazy 放松收敛标准,Conver选项给出更多的控制。
6. 一些程序通过减小积分精度加速SCF。
对于使用弥散函数,长程作用或者低能量激发态的体系,必须使用高积分精度:SCF=NoVarAcc。
7. 尝试改变结构。
首先略微减小键长,接下来略微增加键长,接下来再对结构作一点改变。
8. 考虑使用不同的基组。
9. 考虑使用不同理论级别的计算。
这并不总是实用的,但除此之外,增加迭代数量总是使得计算时间和使用更高理论级别差不多。
10. 关闭DIIS外推(SCF=NoDIIS)。
同时进行更多的迭代( SCF=(MaxCycle=N) )。
11. 更多的SCF迭代( SCF(MaxCycle=N),其中N是迭代数)。
这很少有帮助,但值得一试。
12. 使用强制的收敛方法。
SCF=QC通常最佳,但在极少数情况下SCF=DM 更快。
不要忘记给计算额外增加一千个左右的迭代。
应当测试这个方法获得的波函,保证它最小,并且正好不是稳定点(使用Stable关键字)。



gaussian教程节译自Exploring Chemistry with Electronic Structure Methos, Second Edition,作者James B. Foresman, Eleen Frisch 出版社Gaussian, Inc, USA, 1996前言Gaussian可以做很多事情,具体包括分子能量和结构研究过渡态的能量和结构研究化学键以及反应的能量分子轨道偶极矩和多极矩原子电荷和电势振动频率红外和拉曼光谱核磁极化率和超极化率热力学性质反应途径计算可以模拟在气相和溶液中的体系,模拟基态和激发态.Gaussian是研究诸如取代效应,反应机理,势能面和激发态能量的有力工具.全书结构序言运行Gaussian第一部分基本概念和技术第一章计算模型第二章单点能计算第三章几何优化第四章频率分析第二部分计算化学方法第五章基族的影响第六章理论方法的选择第七章高精度计算第三部分应用第八章研究反应和反应性第九章激发态第十章溶液中的反应附录A 理论背景附录B Gaussian输入方法简介运行GaussianUnix/Linux平台:运行gaussian前要设置好运行参数,比如在C Shell中,需要加这两句setenv g94root directory / directory指程序的上级目录名source $g94root/g94/bsd/g94.login然后运行就可以了.比如有输入文件,采用C Shell时的运行格式是g94 h2o.logWindows平台:图形界面就不用多说了输入输出文件介绍在Unix系统中,输入文件是.com为扩展名的,输出文件为.log;在Windows系统中,输入文件是.gjf为扩展名,输出文件为.out.下面是一个输入文件#T RHF/6-31G(d) TestMy first Gaussian job: water single point energy0 1O -0.464 0.177 0.0H -0.464 1.137 0.0H 0.441 -0.143 0.0第一行以#开头,是运行的说明行,#T表示指打印重要的输出部分,#P表示打印更多的信息.后面的RHF表示限制性Hartree-Fock方法,这里要输入计算所选用的理论方法6-31G(d)是计算所采用的基组,就是用什么样的函数组合来描述轨道Test是指不记入Gaussian工作档案,对于单机版没有用处.第三行是对于这个工作的描述,写什么都行,自己看懂就是了.第二行是空行,这个空行以及第四行的空行都是必须的.第五行的两个数字,分别是分子电荷和自旋多重度.第六行以后是对于分子几何构性的描述.这个例子采用的是迪卡尔坐标.分子结构输入完成后要有一个空行.对于Windows版本,程序的图形界面把这几部分分得很清楚.输入的时候就不要再添空行了.输出文件输出文件一般很长,对于上面的输入文件,其输出文件中,首先是版权说明,然后是作者,Pople的名字在最后一个.然后是Gaussian读入输入文件的说明,再将输入的分子坐标转换为标准内坐标,这些东西都不用去管.当然,验证自己的分子构性对不对就要看这个地方.关键的是有SCF Done的一行,后面的能量可是重要的,单位是原子单位,Hartree,1 Hartree= 4.3597482E-18 Joules或=2625.500 kJ/mol=27.2116 eV再后面是布居分析,有分子轨道情况,各个轨道的本征值(能量),各个原子的电荷,偶极距.然后是整个计算结果的一个总结,各小节之间用\分开,所要的东西基本在里面了.然后是一句格言,随机有Gaussian程序从它的格言库里选出的(在l9999.exe中,想看的可以用文本格式打开这个文件,自己去找,学英语的好机会).然后是CPU时间,注意这不是真正的运行时间,是CPU运行的时间,真正的时间要长一些.如果几个工作一起做的话(Window下好像不可能,Unix/Linix下可以同时做多个工作),实际计算时间就长很多了.最后一句话,"Normal termination of Gaussian 94"很关键,如果没有这句话,说明工作是失败的,肯定在什么地方出错误了.这是这里应该有出错信息.根据输入文件的设置,输出文件还要多一些内容,上面的是基本的东西.第一章计算模型1.1 计算化学的方法主要有分子理论(Molecular Mechanics)和电子结构理论(Electronic Structure Theory).两者的共同点是1. 计算分子的能量,分子的性质可以根据能量按照一定的方法得到.2. 进行几何优化,在起始结构的附近寻找具有最低的能量的结构.几何优化是根据能量的一阶导数进行的.3. 计算分子内运动的频率.计算依据是能量的二阶导数.1.2 分子理论分子理论采用经典物理对分子进行处理,可以在MM3,HyperChem, Quanta, Sybyl, Alchemy等软件中看到.根据所采用的力场的不同,分子理论又分为很多种.分子理论方法很便宜(做量化的经常用贵和便宜来描述计算,实际上就是计算时间的长短,因为对于要花钱上机的而言,时间就是金钱;对于自己有机器的,要想算的快,也要多在机器上花钱),可以计算多达几千个原子的体系.其缺点是1. 每一系列参数都是针对特定原子得出的.没有对于原子各个状态的统一参数.2. 计算中忽略了电子,只考虑键和原子,自然就不能处理有很强电子效应的体系, 比如不能描述键的断裂.1.3 电子结构理论这一理论基于薛定鄂方程,采用量子化学方法对分子进行处理.主要有两类:1. 半经验方法,包括AM1, MINDO/3, PM3,常见的软件包有MOPAC, AMPAC, HyperChem, 以及Gaussian.半经验方法采用了一些实验得来的参数,来帮助对薛定鄂方程的求解.2. 从头算.从头算,在解薛定鄂方程的过程中,只采用了几个物理常数,包括光速,电子和核的质量,普朗克常数,在求解薛定鄂方程的过程中采用一系列的数学近似,不同的近似也就导致了不同的方法.最经典的是Hartree-Fock方法,缩写为HF.从头算能够在很广泛的领域提供比较精确的信息,当然计算量要比前面讲的方法大的多,就是贵得多了.1.4 密度泛函(Density Functional Methods)密度泛函是最近几年兴起的第三类电子结构理论方法.它采用泛函(以函数为变量的函数)对薛定鄂方程进行求解,由于密度泛函包涵了电子相关,它的计算结果要比HF方法好,计算速度也快.1.5 化学模型(Model Chemistries)Gaussian认为所谓理论是,一个理论模型,必须适用于任何种类和大小体系,它的应用限制只应该来自于计算这里包括两点,1. 一个理论模型应该对于任何给定的核和电子有唯一的定义,就是说,对于解薛定鄂方程来讲,分子结构本身就可以提供充分的信息.2. 一个理论模型是没有偏见的,指不依靠于任何的化学结构和化学过程.这样的理论可以被认为是化学理论模型(theoretical-model chemistry),简称化学模型(model chemistry)(这个翻译我可拿不准,在国内没听说过).1.6 定义化学模型Gaussian包含多种化学模型,比如计算方法Gaussian关键词方法HF Hartree-Fock自恰场模型B3L YP Becke型3参数密度泛函模型,采用Lee-Yang-Parr泛函MP2 二级Moller-Plesset微扰理论MP4 四级Moller-Plesset微扰理论QCISD(T) 二次CI具体在第六章讨论基组基组是分子轨道的数学表达,具体见第五章开壳层,闭壳层指电子的自旋状态,对于闭壳层,采用限制性计算方法,在方法关键词前面加R对于开壳层,采用非限制性计算方法,在方法关键词前面加U.比如开壳层的HF就是UHF.对于不加的,程序默认为是闭壳层.一般采用开壳层的可能性是1. 存在奇数个电子,如自由基,一些离子2. 激发态3. 有多个单电子的体系4. 描述键的分裂过程模型的组合高精度的计算往往要几种模型进行组合,比如用中等算法进行结构优化,然后用高精度算法计算能量.第二章单点能计算2.1 单点能计算是指对给定几何构性的分子的能量以及性质进行计算,由于分子的几何构型是固定不变的,只是"一个点",所以叫单点能计算.单点能计算可以用于:计算分子的基本信息可以作为分子构型优化前对分子的检查在由较低等级计算得到的优化结果上进行高精度的计算在计算条件下,体系只能进行单点计算单点能的计算可以在不同理论等级,采用不同基组进行,本章的例子都采用HF方法2.2 计算设置计算设置中,要有如下信息:计算采用的理论等级和计算的种类计算的名称分子结构方法设置这里设置了计算要采用的理论方法,采用的基组,所要进行的计算的种类等信息.这一行,以#开头,默认的计算种类为单点能计算,关键词为SP,可以不写.这一部分需要出现的关键词有,计算的理论,如HF(默认关键词,可以不写),B3PW91;计算采用的基组,如6-31G, Lanl2DZ;布局分析方法,如Pop=Reg;波函数自恰方法,如SCF=Tight.Pop=Reg只在输出文件中打印出最高的5条HOMO轨道和最低的5条LOMU轨道,而采用Pop=Full则打印出全部的分子轨道.SCF设置是指波函数的收敛计算时的设定,一般不用写,SCF=Tight设置表示采用比一般方法较严格的收敛计算.计算的名称一般含有一行,如果是多行,中间不能有空行.在这里描述所进行的计算.分子结构首先是电荷和自旋多重度电荷就是分子体系的电荷了,没有就是0,自旋多重度就是2S+1,其中S是体系的总自旋量子数,其实用单电子数加1就是了.没有单电子,自旋多重度就是1.然后是分子几何构性,一般可以用迪卡尔坐标,也可以用Z-矩阵(Z-Matrix)多步计算Gaussian支持多步计算,就是在一个输入文件中进行多个计算步骤.2.3 输出文件中的信息例2.1 文件e2_01 甲醛的单点能标准几何坐标.找到输出文件中Standard Orientation一行,下面的坐标值就是输入分子的标准几何坐标.能量找到SCF Done: E(RHF)= -113.863697598 A. U. after 6 cycles这里的树脂就是能量,单位是hartree.在一些高等级计算中,往往有不止一个能量值,比如下一行E2=-0.3029540001D+00 EUMP2=-0.11416665769315D+03这里在EUMP2后面的数字是采用MP2计算后的能量.MP4计算的能量输出就更复杂了分子轨道和轨道能级对于按照计算设置所打印出的分子轨道,列出的内容包括,轨道对称性以及电子占据情况,O表示占据,V表示空轨道;分子轨道的本征值,也就是分子轨道的能量,分子轨道的顺序就是按照能量由低到高的顺序排列的;每一个原子轨道对分子轨道的贡献.这里要注意轨道系数,这些数字的相对大小(忽略正负号)表示了组成分子轨道的原子轨道在所组成的分子轨道中的贡献大小.寻找HOMO和LUMO轨道的方法就是看占据轨道和非占据轨道的交界处.电荷分布Gaussian采用的默认的电荷分布计算方法是Mullikin方法,在输出文件中寻找Total atomic charges可以找到分子中所有原子的电荷分布情况.偶极矩和多极矩Gassian提供偶极矩和多极矩的计算,寻找Dipole momemt (Debye),下面就是偶极矩的信息,再下两行是四极矩偶极矩的单位是德拜CPU时间和其他Job cpu time : 0days 0 hours 0 minuites 9.1 seconds.这里是计算的时间,注意是CPU时间.2.4 核磁计算例2.2 文件e2_02 甲烷的核磁计算核磁是单点能计算中另外一个可以提供的数据,在计算的工作设置部分,就是以#开头的一行里,加入NMR关键词就可以了,如#T RHF/6-31G(d) NMR Test在输出文件中,寻找如下信息GIAO Magnetic shielding tensor (ppm)1 C Isotropic = 199.0522 Anisotropy = 0.0000这是采用上面的设置计算的甲烷的核磁结果,所采用的甲烷构形是用B3L YP密度泛函方法优化得到的.一般的,核磁数据是以TMS为零点的,下面是用同样的方法计算的TMS(四甲基硅烷)的结果1 C Isotropic = 195.1196 Anisotropy = 17.5214这样,计算所得的甲烷的核磁共振数据就是-3.9ppm,与实验值-7.0ppm相比,还是很接近的.2.5 练习练习2.1 文件2_01 丙烷的单点能练习要点:寻找分子的标准坐标,寻找单点能,偶极矩的方向和大小,电荷分布练习2.2 文件2_02a (RR), 2_02b (SS), 2_02c (RS) 1,2-二氯-1,2-二氟乙烷的能量练习要点:比较该化合物三个旋光异构体的能量和偶极矩差异练习2.3 文件2_03 丙酮和甲醛的比较练习要点:比较甲基取代氢原子后带来的影响说明能量比较必须在有同样的原子种类和数量的情况下进行练习2.4 文件2_04 乙烯和甲醛的分子轨道练习要点:寻找HOMO和LUMO能级,并分析能级的组成情况练习2.5 文件2_05a, 2_05b, 2_05c 烷,烯,炔的核磁共振比较练习2.6 文件2_06 C60的单点能练习要点:分析C60最高占据轨道注意在收敛方法选择的时候,要有SCF=Tight,否则有收敛问题.练习2.7 文件2_07 计算大小的CPU资源比较本练习比较不同基组函数数量,SCF方法对CPU时间,资源的占用情况.比较传统SCF方法(SCF=Convern),直接SCF方法(Gaussian默认方法)传统SCF 直接SCF基组函数数量int文件大小(MB) CPU时间CPU时间23 2 8.6 12.842 4 11.9 19.861 16 23.2 38.880 42 48.7 72.199 92 95.4 122.5118 174 163.4 186.8137 290 354.5 268.0156 437 526.5 375.0175 620 740.2 488.0194 832 1028.4 622.1很显然,函数数量对资源占用和CPU时间都有很大影响,函数越多,资源占用越大,CPU时间越长.理论上来讲,认为CPU时间和函数数量的四次方成正比,但实际上没有这么高, 在本例中,基本上和函数数量的2.5次方成正比.一般的讲,直接SCF方法的效率要比传统SCF方法要好,在本例中,当函数数量比较大时, 可以看到这一点.练习2.8 文件2_08a (O2), 2_08b (O3) SCF稳定性计算本例中采用SCF方法分析分子的稳定性.对于未知的体系,SCF稳定性是必须要做的.当分子本身不稳定的时候,所得到的SCF结果以及波函数等信息就没有化学意义.SCF稳定性分析是寻找是不是存在比当前状态能量更低的分子状态.关键词有Stable 检验分子的稳定性,放松对分子的限制,比如由闭壳层改为开壳层等.Stable=OPT 这一选项设定,当发现不稳定的时候,对新的状态进行优化.这种做法一般是不推荐的,因为所得到的新的状态的几何形太接近原来的几何构形.本例中首先计算闭壳层的单重态的氧分子.很显然,闭壳层单重态的氧分子不应该是稳定的.在输出文件中,我们可以找到这样的句子:The wavefuction has an RHF --> UHF instability.这表明存在一个UHF的状态,其能量要比当前状态低.这说明可能,能量最低的状态是单重态的,但不是闭壳层的;存在有更低能量的三重态;所计算的状态不是能量最低点,可能是过渡态.在三重态情况下重新计算,也进行稳定性验证,可以看到如下的句子The wavefunction is stable under the perturbations considered.臭氧是单重态的,但有不一般的电子结构.采用RHF Stable=Opt可以发现一个RHF-->UHF的不稳定性,在所得到的UHF状态下进行稳定性检验,采用UHF Stable=Opt,发现体系仍然不稳定.The wavefunction has an inernal instability再在此基础上进行的优化,体系又回到了RHF的状态.这时,就需要在进行SCF前的构性初始电子状态猜测上进行改动,使用Guess=Mix,在初始猜测中混合HOMO 和LUMO轨道,从而消除空间对称性,然后进行的UHF Guess=Mix Stable 表明得到了稳定的结构.确定电子状态还可以采用Guess=Alter详见Gaussian User's Reference第三章几何优化前面讨论了在特定几何构型下的能量的计算,可以看出,分子几何构型的变化对能量有很大的影响.由于分子几何构型而产生的能量的变化,被称为势能面.势能面是连接几何构型和能量的数学关系.对于双原子分子,能量的变化与两原子间的距离相关,这样得到势能曲线,对于大的体系,势能面是多维的,其维数取决与分子的自由度.3.1势能面势能面中,包括一些重要的点,包括全局最大值,局域极大值,全局最小值,局域极小值以及鞍点.极大值是一个区域内的能量最高点,向任何方向的几何变化都能够引起能量的减小.在所有的局域极大值中的最大值,就是全局最大值;极小值也同样,在所有极小之中最小的一个就是具有最稳定几何结构的一点.鞍点则是在一个方向上具有极大值,而在其他方向上具有极小值的点.一般的,鞍点代表连接着两个极小值的过渡态.寻找极小值几何优化做的工作就是寻找极小值,而这个极小值,就是分子的稳定的几何形态.对于所有的极小值和鞍点,其能量的一阶导数,也就是梯度,都是零,这样的点被称为稳定点.所有的成功的优化都在寻找稳定点,虽然找到的并不一定就是所预期的点. 几何优化有初始构型开始,计算能量和梯度,然后决定下一步的方向和步长,其方向总是向能量下降最快的方向进行.大多数的优化也计算能量的二阶导数,来修正力矩阵,从而表明在该点的曲度.3.2 收敛标准当一阶导数为零的时候优化结束,但实际计算上,当变化很小,小于某个量的时候,就可以认为得到优化结构.对于Gaussian,默认的条件是力的最大值必须小于0.00045,均方根小于0.0003为下一步所做的取代计算为小于0.0018,其均方根小于0.0012这四个条件必须同时满足,比如,对于非常松弛的体系,势能面很平缓,力的值已经小于域值,但优化过程仍然有很长的路要走.对于非常松弛的体系,当力的值已经低于域值两个数量级,尽管取代计算仍然高于域值,系统也认为找到了最优点.这条规则用于非常大,非常松弛的体系.3.3 几何优化的输入Opt关键字描述了几何优化例3.1 文件e3_01 乙烯的优化输入文件的设置行为#R RHF/6-31G(d) Opt Test表明采用RHF方法,6-31G(d)基组进行优化3.4 输出文件优化部分的计算包含在两行相同的GradGradGradGradGradGradGradGradGradGrad...........之间,这里有优化的次数,变量的变化,收敛的结果等等.注意这里面的长度单位是波尔.在得到每一个新的几何构型之后,都要计算单点能,然后再在此基础上继续进行优化,直到四个条件都得到满足.而最后一个几何构型就被认为是最优构型.注意,最终构型的能量是在最后一次优化计算之前得到的.在得到最优构型之后,在文件中寻找--Stationmay point found.其下面的表格中列出的就是最后的优化结果以及分子坐标.随后按照设置行的要求,列出分子有关性质例3.2 文件e3_02 氟代乙烯的优化3.5 寻找过渡态Gaissian使用STQN方法确定反应过渡态,关键词是Opt=QST2例3.3 文件e3_03 过渡态优化例中分析的是H3CO --> H2COH 的变化,输入文件格式#T UHF/6-31G(d) Opt=QST2 TestH3CO --> H2COH Reactants0,2structure for H3CO0,2structure for H2COHGaussian也提供QST3方法,可以优化反应物,产物和一个由用户定义的猜测的过渡态.3.6 难处例的优化有一些系统的优化很难进行,采用默认的方法得不到结果,其产生的原因往往是所计算出的力矩阵与实际的相差太远.当默认方法得不到结果时,就要采用其他的方法. Gaussian提供很多的选择,具体可以看User's Reference.下面列举一些.Opt=ReadFC 从频率分析(往往是采用低等级的计算得到的)所得到的checkpoint文件中读取初始力矩阵,这一选项需要在设置行之前加入%Chk= filename 一句,说明文件的名称.Opt=CalCFC 采用优化方法同样的基组来计算力矩阵的初始值.Opt=CalcAll 在优化的每一步都计算力矩阵.这是非常昂贵的计算方法,只在非常极端的条件下使用.有时候,优化往往只需要更多的次数就可以达到好的结果,这可以通过设置MaxCycle 来实现.如果在优化中保存了Checkpoint文件,那么使用Opt=Restart可以继续所进行的优化.当优化没有达到效果的时候,不要盲目的加大优化次数.这是注意观察每一步优化的区别,寻找没有得到优化结果的原因,判断体系是否收敛,如果体系能量有越来越小的趋势,那么增加优化次数是可能得到结果的,如果体系能量变化没有什么规律,或者,离最小点越来越远,那么就要改变优化的方法.也可以从输出文件的某一个中间构型开始新的优化,关键词Geom=(Check,Step=n)表示在取得在Checkpoint文件中第n步优化的几何构型3.7 练习练习3.1 文件3_01a (180), 3_01b (0) 丙烯的优化从两种丙烯的几何异构体进行优化,一个是甲基的一个氢原子与CCH形成180度二面角,另一个是0.优化结果表明,二者有0.003Hartree的差别,0度的要低.练习3.2 文件3_02a (0), 3_02b (180), 3_02c (acteald.) 乙烯醇的优化乙烯醇氧端的氢原子与OCC平面的二面角可以为0和180,优化得到的结果时,0度的能量比180度的低0.003Hartree,但同时做的乙醛的优化表明,乙醛的能量还要低,比0度异构体低0.027hartree.练习3.3 文件3_03 乙烯胺的优化运行所有原子都在同一平面上的乙烯胺的优化.比较本章的例子和练习,可以看到不同取代基对乙烯碳碳双键的影响.练习3.4 文件3_04 六羰基铬的优化本例采用STO-3G和3-21G基组,在设置行中加入SCF=NoVarAcc对收敛有帮助.3-21G基组的优化结果要优于STO-3G练习3.5 文件3_05a (C6H6), 3_05b (TMS) 苯的核磁共振采用6-31G(d)基组,B3L YP方法优化几何构性,采用HF方法,6-311+G(2d,p)基组在优化的几何构型基础上计算碳的化学位移.注意,核磁共振的可靠程度依赖准确的几何结构和大的基组.输入文件如下%Chk=NMR#T B3L YP/6-31G(d) Opt TestOptmolecule specification--Link1--%Chk=NMR%NoSave#T RHF/6-311+G(2d,p) NMR Geom=Check Guess=Read TestNMRcharg & spin同样,还需要采用同样方法计算TMS.下面是计算结果绝对位移相对位移实验值TMS Benzene188.7879 57.6198 131.2 130.9练习3.6 文件3_06a (PM3), 3_06b (STO-3G) 氧化碳60的优化C60中有两种碳碳键,一是连接两个六元环的6-6键,另一是连接六元环和无元环的5-6键. 氧化C60就有两种异构体.本例采用PM3和HF/STO-3G方法来判断那种异构体是稳定的,以及氧化后的C-C键的变化.采用Opt=AddRedundant关键词可以在输出文件中打印所要求的键长,键角,这一关键词需要在分子构型输入结束后在增加关于所要键长键角的信息,键长用两个原子的序列号表示,键角则用三个原子表示.计算结果显示,6-6键的氧化,碳碳键仍然存在,接近环氧化合物,而5-6键已经打开.采用不同的方法,得到的几何结构相差不多,但在能量上有很大差异.在采用MNDO,PM3,HF/3-21G方法得到的能量数据中,5-6键氧化的异构体的能量低,但采用HF/STO-3G得到的结果,确实6-6键氧化的能量低.Raghavachari在其进行的上述研究中阐述动力学因素同样是重要的;实验上还没有发现那个是能量最低的异构体;应该进行更精确的计算练习3.7 文件3_07 一个1,1消除反应的过渡态优化分析反应SiH4 --> SiH2 + H2, 可以采用Opt=(QST2, AddRedundant)关键词来进行过渡态优化,同时特别关注过渡态结构中的某个键长练习3.8 文件3_08 优化进程比较采用下述三种方法优化二环[2,2,2]直接采用默认方式冗余内坐标优化Opt;采用迪卡尔坐标优化Opt=Cartesian;采用内坐标优化Opt=Z-Matrix结果显示,冗余内坐标优化的优化次数最短,内坐标优化的次数最多.第四章频率分析频率分析可以用于多种目的,预测分子的红外和拉曼光谱(频率和强度)为几何优化计算力矩阵判断分子在势能面上的位置计算零点能和热力学数据如系统的熵和焓4.1 红外和拉曼光谱几何优化和单点能计算都将原子理想化了,实际上原子一直处于振动状态.在平衡态,这些振动是规则的和可以预测的.频率分析的计算要采用能量对原子位置的二阶导数.HF方法,密度泛函方法(如B3L YP), 二阶Moller-Plesset方法(MP2)和CASSCF方法(CASSCF)都可以提供解析二阶导数.对于其他方法,可以提供数值二阶导数.4.2 频率分析输入Freq关键词代表频率分析.频率分析只能在势能面的稳定点进行,这样,频率分析就必须在已经优化好的结构上进行.最直接的办法就是在设置行同时设置几何优化和频率分析.特别注意的是,频率分析计算是所采用的基组和理论方法,必须与得到该几何构型采用的方法完全相同!例4.1 文件e4_01 甲醛的频率分析例中采用的是已经优化好的几何构型,输入格式# RHF/6-31G(d) Freq Test4.3 频率和强度频率分析首先要计算输入结构的能量,然后计算频率.Gaussian提供每个振动模式的频率,强度,拉曼极化率.以下是例4.1的输出文件中的前四个频率1 2 3 4B1 B2 A1 A1。



一Gaussian03运行1.1Scratch文件Gaussian运行中使用数个Scratch文件,包括:CheckPoint文件:name.chk读写文件:name.rwf双电子积分文件:name.int双电子积分的导数文件name.d2e默认情况下这些文件由Gaussian处理进程ID命名并储存于Scratch中,计算结束后删除。
一般情况下我们希望保存CheckPoint 文件,此时只需要在输入文件中给CheckPoint 文件命名或者指定路径即可。
常用格式:%Chk=name;%Chk=chem/scratch/name。
%RWF=path读写文件%Int=path 积分文件%D2E=path积分导数文件通过以上三个命令可以将以上三个文件分别存入不同的盘中。
%NoSave命令%RWF=/chem/scratch2/water到这里为止的文件都被删除%NoSave%Chk=water到这里为止的文件都被保存1.2控制内存使用%mem=***KB/MB/GB/GW二Gaussian03的输入2.1Gaussian03的输入文件概述Gaussian03输入文件的基本结构如下Gaussian03输入界面Gaussian03的输入部分分类任务类型计算方法基组输入语法Gaussian语法规则:输入是自由格式,而且与大小写无关;空格、TAB、正斜线/、逗号都可以作为一行之内的不同项之间的连接符。
关键字格式:keyword = 选项keyword(选项)keyword=(选项1, 选项2, ...)keyword(选项1, 选项2, ...)这些选项中可以带数值。
在Gaussian中所有的关键字和选项都必须使用可以识别的缩写。
引用外部文件使用@文件名。
注释行以!开始。
Gaussian03任务类型形式为方法2/基组2 // 方法1/基组1 的计算执行路径,表示用方法1/基组1进行几何优化计算,之后在优化的结构上用方法2/基组2进行单点能计算。

Technical Support InformationLast update: 16 April 2003Overlay 15 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 88 89 90 91 92 94 95 96 97 98 101 102 103 104 105 106 107 108 109 110 111 112 113 114Overlay 29 10 11 12 13 14 15 16 17 18 19 20 29 30 40 41Overlay 35 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 67 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106Overlay 45 6 7 8 9 10 11 13 14 15 16 17 18 19 20 21 22 23 24 25 26 28 29 31 33 34 35 36 37 38 43 44 45 46 47 48 60 61 62 63 64 65 66 67 68 69 71 72 80 81 82 110Overlay 55 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102Overlay 615 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 70 71 72 73 74 75 76 77 78 79 80 81 82 83Overlay 76 7 8 9 10 11 12 13 14 15 16 18 25 29 30 31 32 40 41 42 43 44 45 52 53 64 65 70 71 72 74 75 76 77 87Overlay 85 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 27 28 29 30 31 32 35 36 38 39 40 41 42 43 44 45 46 47Overlay 95 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 25 26 27 28 30 31 36 37 38 40 41 42 43 44 45 46 47 48 49 60 61 62 70 71 72 73 74 75 81 82 83 84 85 86Overlay 105 6 7 8 9 10 11 13 14 15 16 17 18 19 20 21 22 28 29 30 31 32 45 46 47 48 60 61 62 63 72 72 74 75 76 77 78 79Overlay 115 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 26 27 28 29 30 31 32 33 39 40 41 42 43 45 46 53 60 61 62 63 70 71 75Overlay 99995 6 7 8 9 10 11 12 13 14 15 16 17 18 33Technical Support InformationLast update: 22 March 2003Overlay 15 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 88 89 90 91 92 94 95 96 97 98 101 102 103 104 105 106 107 108 109 110 111 112 113 114IOp(1/5)L103 MODE OF OPTIMIZATION 0 FIND LOCAL MINIMUM 1 FIND A SADDLE POINT N FIND A STATIONARY POINT ON THE ENERGY SURFACE WITH N NEGATIVE EIGENVALUES OF THE 2ND DERIVATIVE MATRIX L107: MODE OF SEARCH 0 LOCATE THE MAXIMUM IN THE LST PATH. 1 SCAN THE LST PATH.IOp(1/6)L102, L103, L105, L107, L109, L112, L113, L114: MAXIMUM NUMBER OF STEPS (OR NUMBER OF STEPS FOR AN LST SCAN). 0 NSTEP = Max(20,NVAR+10) (L103, L112) = Min(20,NVAR+10) (L102, L105, L109) = Min(40,NVar+20) (L113, L114) N NSTEP = NIOp(1/7)L103, L105, L109, L112, L113, L114: CONVERGENCE ON THE FIRST DERIVATIVE AND ESTIMATED DISPLACEMENT FOR THE OPTIMIZATION RMS FIRST DERIVATIVE .LT. CONFV, RMS EST. DISPLACEMENT .LT. CONVX=4*CONVF -1 ConvF = 1/600 HARTREE/BOHR OR RADIAN 0 CONVF = 0.0003 HARTREE/BOHR OR RADIAN N CONVF = N*10**-6 L116, L117: Convergence on electric field/charges -1 Default value for optimizations: 10**-7. 0 Default value for single-points: 10**-5 in L116, 10**-7 in L117. N 10**-N.IOp(1/8)L103, L109, L112: MAXIMUM STEP SIZE ALLOWED DURING OPT. 0 DXMAXT = 0.1 BOHR OR RADIAN (L103, Estm or UnitFC). = 0.3 Bohr or Radian (L103, Read or CalcFC). = 0.2 Bohr or Radian (L105). = 0.3 Bohr or Radian (L113, L114). DXMAXT = 0.01 * NNL117: General control. 0 Which type of basin to use to partition the density isosurface. Default is 4 1 GradVne 2 GradRho 3 Don't Use Basins, Use only the Center of NuclearCharge 4 Use Interlocking Spheres N0 Order of Adam's-Bashforth-Moulton (ABM) predictorcorrector method to use in solving diff. eqns. for the grad RHO or Vne trajectories. Default is 4, max is 9. N00 Number of small steps per ABM step to be used in starting ABM and when "slow down" is needed in ABM. Default is 5. N000 Which approximation to make. Default is III for Tomasi (interlocking spheres) and IV for general surface. 1000 Apprx. I - Don't Do Self-Polarization or "Compensation" 2000 Apprx. II - Do-Self Polarization, But No Compensation. 3000 Apprx. III - Do Self-Polarization and Compensation. 4000 Apprx. IV - Do III and Allow Surface To "Relax" in Solution if no spheres N0000 Whether to evaluate densities using orbitals or density matrix. Default is to use density. 10000 Use MOs. 20000 Use density. L121: Time step, N*0.0001 fs, default 0.1IOp(1/9)L103: Use of Trust radius. 0 Whether to update trust radius (DXMaxT, default Yes). Default is Yes for minima, no for TS. 1 2 No. Yes.00 Whether to scale or search the sphere when reducing the step size to the trust radius (Default search for minima, scale for transition states.). 10 20 Scale. Search.L107: WHETHER TO MAINTAIN SYMMETRY ALONG THE SEARCH PATH. 0 1 YES. NO.L117: Whether to delete points which are too close together: 0 1 No Yes, using a default criteria (0.05 Angstroms)-N Yes, using a (10**-N Angstroms) criteria. How close to get to the isosurface in search. 0 N Approx 1.0D-6 (N=20) 2.0**-NL121: Whether to read in initial velocities: 0 1 2 3 Default (same as 1) Generate random initial velocity Read in initial cartesian velocity (Bohr/sec) Read in initial MW cartesian velocity (sqrt(amu)*Bohr/sec)IOp(1/10)L103, L105, L109, L112, L113, L114: Input of initial Hessian: All values must be in atomic units (Hartree, Bohr, and radians). 0 1 2 3 Use defaults (not valid for L109). Read ((FC(I,J),J=1,I),I=1,NVAR) (8F10.6) (L103 only). Read I,J,FC(I,J), (5I3,F20.0) (L103 only). End with a blank card. Read from checkpoint file in internal coordinates.4 Second derivative matrix calculated analytically. (not valid for L109). 5 Read cartesian forces and force constants from the checkpoint file are convert to internal coordinates. 6 Read cartesian forces followed by cartesian force constants (both in format 6F12.8) from input stream. followed by a blank line. 7 8 9 10 Use semiempirical force constants. Use unit matrix (default for L105; only recognized by 103). Estimate force constants using valence force field. Use unit matrix throughout.IOp(1/11)L103: TEST OF CURVATURE. BOMB THE JOB IF THE SECOND SECOND DERIVATIVE MATRIX HAS THE WRONG NUMBER OF NEGATIVE EIGENVALUES. 0 DEFAULT (TEST for z-matrix or cartesian TS but not for LST/QST or for minimum). 1 2 DON'T TEST. TEST.L117: Scaling Factor for Determining Overlaps of VDW atoms -1 0 N Turn off scaling Default is 1.010 1.000 + N*(0.001)Step size for ABM method in Trudge for isodensity method. 0 N 0.05 (N=2) 0.1/NIOp(1/12)L103: OPTIMIZATION CONTROL PARAMETERS 0 USE DEFAULT VALUES1 READ IN NEW VALUES FOR ALL PARAMETERS (SEE INITBS)IOp(1/13)L103,L113,L114,L115: Type of Hessian Update: 0 Default (9 for L103 minimization, 7 for L103 TS, D2Corr and L115, Powell for L113 and L114). 1 2 3 4 5 6 7 8 9 Powell (not in L103). BFGS (not in L103) BFGS, safeguarding positive definateness (not inL103 or L115) D2Corr (New, only in L103 and L115). D2Corr (Old, only in L103 and L115). D2Corr (BFGS) D2Corr (Bofill Powell+MS for transition states). D2Corr (No update, use initial Hessian). D2Corr (New if energy rises, otherwise BFGS).L121: Multi-time step parameter (NDtrC,NDtrP) 0 NN No multi-time stepping Iterate density constraints NN times per stepMM00 Do gradient once every MM stepsIOp(1/14)L103: Max. number of bad steps to allow before attempting a linear minimization (i.e., no quadratic step). 0 N Default (0 for TS, 1 for minima). Allow N -- linear only starts with the N+1st.IOp(1/15)L103,L109: ABORT IF DERIVATIVES TOO LARGE -1 or 0 N No force test at all. FMAXT = 0.1 * NIOp(1/16)L103,L113,L114: MAXIMUM ALLOWABLE MAGNITUDE OF THE EIGENVALUESOF THE SECOND DERIVATIVE MATRIX. IF THE LIMIT IS EXCEEDED, THE SIZE OF THE EIGENVALUE IS REDUCED TO THE MAXIMUM, AND PROCESSING CONTINUES. 0 N EIGMAX = 25.0 HARTREE / BOHR**2 OR RADIAN**2 EIGMAX = 0.1 * NIOp(1/17)L103,L113,L114: MINIMUM ALLOWABLE MAGNITUDE OF THE EIGENVALUES OF THE SECOND DERIVATIVE MATRIX. SIMMILAR TO IOp(16) 0 N EIGMIN = 0.0001 EIGMIN = 1. / NIOp(1/18)L103: Coordinate system. 0 Proceed normally1 Second derivatives will be computed as directed on the variable definition cards. No optimization will occur. 10 20 30 40 Do optimization in cartesian coordinates. Do full optimization in redundant internal coord. Do full optimization in pruned distance matrix coords. Do optimization in Z-matrix coordinates.50 Do full optimization in redundant internal coords with large molecular tools. 100 1000 2000 3000 Read the AddRedundant input section for each structure. Do not define H-bonds Define H-bonds with no related coordinates (default) Define H-bonds and related coordinates10000 Reduce the number of redundant internals 20000 Define all redundant internals 100000 0000000 Old definition of redundant internals. Default (2000000).1000000 Skip MM atoms in internal coordinate definitions and do microiterations the old way, in L103. 2000000 Include MM atoms in internal coordinate definitions (no microiterations). 3000000 Skip MM atoms in internal coordinate definitions and do microiterations the new way, in L120. 4000000 Microiterations for pure MM, done in L402.IOp(1/19)L103: SEARCH SELECTION 0 2 Default (same as 6). LINEAR AND STEEPEST DESCENT.3 STEEPEST DESCENT AND LINEAR ONLY WHEN ESSENTIAL. 4 5 6 7 8 9 10 11 13 Quadratic if curvature is correct; RFO if not. Linear as usual. Quadratic if curvature is correct; RFO if not. No linear search. RFO and linear. RFO without linear. Newton-Raphson and linear. Newton-Raphson only. GDIIS and linear GDIIS only. First-order simultaneous optimization.L113,L114: Search Selection: 0 P-RFO OR RFO STEP ONLY (DEFAULT)1 P-RFO OR RFO STEP FOR "WRONG" HESSIAN OTHERWISE NEWTON-RAPHSONIOp(1/20)L101, L106, L108, L109, L110: INPUT UNITS 0 1 2 3 ANGSTROMS DEGREES BOHRS DEGREESANGSTROMS RADIANS BOHRS RADIANSIOp(1/21)L103,L113,L114: EXPERT SWITCH. 0 NORMAL MODE.1 EXPERT MODE: CERTAIN CUTOFFS USED TO CONTROL THE OPTIMIZATION WILL BE RELAXED. THESE INCLUDE FMAXT, DXMAXT, EIGMAX AND EIGMIN.IOp(1/22)L107: Whether to reorder coordinates for maximum coincidence. 0 1 2 Yes. Assume reactant order equals product order. Read in a re-ordering vector from the input.L115: KIND OF SEARCH: 0 1 2 3 4 5 6 7 BOTH DIRECTIONS AND GENERATE SEARCH VECTOR FORWARD DIRECTION AND GENERATE S. VECTOR BACKWARD DIRECTION AND GENERATE S. VECTOR BOTH DIRECTIONS AND GENERATE S. VECTOR FORWARD DIRECTION AND READ S. VECTOR 8F10.6 FORWARD DIRECTION AND READ S. VECTOR 8F10.6 BACKWARD DIRECTION AND READ S. VECTOR 8F10.6 BOTH DIRECTIONS AND READ S. VECTOR 8F10.6IOp(1/23)L112: Derivative availability. 0 1 2 Energy only. Energy + Forces. Energy + Forces + Force constantsIOp(1/24)Whether to round tetrahedral angles. 0 1 2 Default (Yes). Yes, round angles within 0.001 degree. No.IOp(1/25)Wether SCRF is used with numerical polarizability: 0 1 No. Yes, the field in /Gen/ must be cleared each time.IOp(1/26)Accuracy of function being optimized: -NNMM Energy 10**-(NN), Gradient 10**-(MM). -1 0 1 2 3 Read in values Default (same as 1). Normal accuracy for HF (energy and gradient both 1.d-7). Standard grid accuracy for DFT (Energy 1.d-5, gradient 1.d-4) Fine grid accuracy for DFT (Energy 1.d-7, gradient 1.d-6)IOp(1/27)= IJKL (i.e. 1000*I+100*J+10*K+L) Transition state searching using QST and redundant internal coordinates L= 0,1 Input one structure, either initial guess of the minimizing structure or transition structure without QST. L= 2 Input 2 structures, the first one is the reactant, the second one is the product. The union of the two redundant coordinates are taken as the redundant coords for the TS. The values of the TS coord are estimated by interpolating the sturcture of R and P. R and P are used to guide the QST optimization of the TS. L= 3 Input 3 structures. The first one is reactant the second one is the product. The third one is the initial guess of the transition structure. R and P are used to guide the QST optimization of the TS. K = 1-9 Interpolation of initial guess of TS between R and P (TS=0.1*J*R + 0.1*(10-J)*P, default J=5) J=1 J=2 J=3 J=4 LST constraint in internals QST constraint in internals LST constraint in distance matrix space QST constraint in distance matrix spaceI = 0-9 Control parameters for climbing phase of QST (e.g. QSTRad = 0.01*I, default QSTrad = 0.05)IOp(1/28)L103: Number of translations and rotations to remove during redundant coordinate transformations: -2 -1 0 N 0. Normal (6 or 5 for linear molecules). Default, same as -1. N.IOp(1/29)L101: SPECIFICATION OF NUCLEAR CENTERS 0 BY Z-MATRIX1 BY DIRECT COORDINATE INPUT (must set IOp(29) in L202). 2 GET Z-MATRIX AND VARIABLES FROM THE CHECKPOINT FILE. 3 GET CARTESIAN COORDINATES ONLY FROM THE CHECKPOINT FILE. 4 5 By model builder, model A. By model builder, model B.6 Get Z-matrix from the checkpoint file, but read new values for some variables from the input stream. 7 Get all input (title, charge and multiplicity, structure) from the checkpoint file. 10 000 Print details of the model building process. Default (same as 100).100 Do not abort job if model builder generates a z-matrix with too many variables. 200 Abort job if model builder generates a z-matrix with too many variables. 1000 2000 3000 4000 5000 00000 Read optimization flags in format 50L1 after the z-matrix. Set all optimization flags to optimize. Purge flags except the frozen variables. Rebuild the coordinate system. (2+3) Purge all flags but keep the coordinate definition. Default, same as 10000.10000 Mark Z-matrix constants as frozen variables rather than wired-in constants. 20000 Do not retain symbolic constants.100000 Generate a symbolic z-matrix using all Cartesians if none is present on the checkpoint file (a hack to make IRCs work with Cartesian input). 200000 Same as one, but retain the redudant internal coordinate definitions.IOp(1/30)L103: ARE THE READ-WRITE FILES TO BE UPDATED? THIS OPTION IS SET FOR THE LAST CALL TO 103 IN FREQUENCY CALCULATIONS IN ORDER TO PRESERVE THE VALUES OF THE VARIABLES FOR ARCHIVING. It also suppresses error termination on large gradients. 0 1 YES NOIOp(1/32)TITLE CARD PUNCH CONTROL. 0 1 DON'T PUNCH. PUNCH.IOp(1/33)L101: L102 L103 L106 L109 L110 L113 L114 0 1 OFF ON DEBUG PRINTIOp(1/34)L101 L102 L103: DEBUG + DUMP PRINT 0 1 OFF ONIOp(1/35)RESTART (L102-L112). 0 NORMAL OPTIMIZATION.1 FIRST POINT OF A RESTART. GET GEOMETRY, WAVEFUNCTION, ET. FROM THE CHECKPOINT FILE.IOp(1/36)CHECKPOINT. 0 1 NORMAL CHECKPOINT OF OPTIMIZATION. SUPPRESS CHECKPOINTING.IOp(1/37)D2E CLEANUP (obsolete) 0 NO CLEANUP.1 THIS IS THE LAST POINT AT WHICH ANALYTIC SECOND DERIVATIVES WILL BE DONE. DELETE THE D2E FILE AND THE BUCKETS AND TRUNCATE THE READ/WRITE FILES.IOp(1/38)Entry control option (currently only by L106, L107, L108, L109, L110, L111, and L112 but not L102, L103, and L105). 0 1 N>1 . Continuation of run. Initial entry. In L103: Initial entry of guided optimization using N levels.N0 In L106: differentiate Nth derivatives once. In L110 and L111: differentiate energy N times. 000 100 200 In L106: differentiate wrt nuclear coordinates. In L106: differentiate wrt electric field. In L106: differentiate wrt field and nuclear.IOp(1/39)Step size control for numerical differentiation. (L106, L109, L110, L111). Path step size in L115. 0 Use internal default (0.001 Angstroms in L106, 0.005 A in L109, 0.01 Angstrom in L110, 0.001 au in L111). N Use step-size of 0.0001*N (angstroms in L106, L109, L110, electric field au in L111). -1 Read stepsize (up to 2 for L106, 1 for others), free-format.-N>1 Use step-size of 0.0001*N atomic units everywhere.IOp(1/40)L113, L114: Hessian recalculation. -1 0 N Pick up analytic second derivatives every time. Just update. The default, execpt for CalcAll. Recalculation the Hessian every N steps.L116: Whether to read initial E-field: 0 1 2 Start with 0.0. Read from checkpoint file. Read from input stream.IOp(1/41)Step number of optimization from which to take geometry. -1 for the initial geometryIOp(1/42)L103, L115: Number of points along the reaction path in each direction. Default is 6. L117: Cutoff to be used in evaluating densities. 0 N 1.0D-10 1.0D-NIOp(1/43)L116: Extent of Reaction Field. 0 1 2 3 Dipole Quadrupole Octapole HexadecapoleL117: How to define Radii 0 1 2 10 20 30 Default is 11 Use internally stored Radii, centers will be on atoms Read-in centers and radii on cards Force Merz-Kollman radii (Default) Force CHELP (Francl) recommended radii. Force CHELPG (Breneman) recommended radii.100 Read in replacement radii for selected atom types as pairs (IAn,Rad) or (Symbol,Rad), terminated by a blank line. 200 Read in replacment radii for selected atoms as pairs (I,Rad), terminated by a blank line. Initial radius of spheres to be placed around attractors to "capture" the gradient trajectories. The final radius is then automatically optimized separately for each atom. 0 NM 0.1 N.M = NM/10IOp(1/44)IRC Type 0 1 2 3 Default (same as 3). Cartesian. Internal. Mass-weighted.L117: Maximum distance between a nucleus and its portion of the isosurface - used in Trudge only to eliminate, from the outset, points which clearly lie in another basin. This parameter should be chosen with the parameter Cont in mind 0 NM 10.0 au N.M au = NM/10L121: Seed for random number generator (ISeed) -1 Use system time initialize iseed (Note each run will give different results) 0 N Use default seed value (ISeed = 398465) Set random number seed to NIOp(1/45)Read isotopes in L115. 0 1 Do not read isotopes. Read Isotopes.IOp(1/46)Order of multipoles in numerical SCRF: 0 1 2 3 Dipole Quadrupole Octapole Hexadecapole.IOp(1/47)Number of redundant internal coordinates to allow for. 0 N Default: 50000 N.IOp(1/48)IRCMax control. 1 20 Do IRCMax Include zero-point energy.CIOp(1/49)Options to IRC path relaxation (IJKL) L 2/1 dont/do optimize reactant structure. Default: 1K2/1 dont/do optimize product structure. Default: 1J 3/2/1 dont/QST3/QST2 optimize TS structure (for QST input). Default: 1 I 2/1 unimolecular/bimolecular reaction. Default: unimolecularIOp(1/52)L101 and L120: Type of ONIOM calculation: 0/1 2 3 00 10 20 100 One layer, normal calculation Two layers Three layers Default (20) Include electrostatics in model systems using MM charges. No electrostatics included in the model systems Do full square for testing.N000 Use atomic charge type N-1 during microiterations. The default is MK charges.IOp(1/53)L120: Action of each invocation of L120: 0 1 Do nothing Set up point MM on rwf from initial data2 Set up point MM on rwf from initial data and restore point MM on chk file if ONIOM data is present there. 3 4 5 6 Restore point M from data on the rwf. Integrate energy Integrate energy and gradient Integrate energy, gradient, and hessian7 Restore point MM from RWF but do not create a new model system. NN0 Save necessary information (some rwf's, energy, gradients, hessian) of point NN of the ONIOM grid. NN = MaxLev**2 + 1 (currently 17) to restore real system. MM000 Calc Level High ||| Mid ||| Low SML 1--3--6 system size 2--5--8 4--7--9* Next point to do is MM.IOp(1/54)Whether to recover initial energy during IRCMax from chk file: 0 1 No. Yes.IOp(1/55)L103: Options for GDIIS: ICos*1000+IChkC*100+IMix*10+Method form. L115: IRC optimization. 0 1 Default, use gradients to find the next geometry. Use displacements to find the next geometry.IOp(1/56)Set of atom type names to parse: 0 1 2 Accept any. Dreiding/UFF. Amber.3 Amber allowing any symbol, for use with parameters in input stream.IOp(1/57)Whether to produce connectivity: 0 Default (4 if reading geom from chk file and connectivity is there, otherwise 3). 1 2 3 4 5 10 No. Yes, read from input stream Yes, generate connectivity. Yes, read from checkpoint file. Yes, read from rwf file. Read modifications.100 Connectivity input is in terms of z-matrix entries, including dummy atoms.IOp(1/58)IRCMax control in L115.IOp(1/59)Update of coordinates in L103 0 1 2 Default (1 for large opt, 2 for regular) New versions. Old version.IOp(1/60)Interpret extra integer and fp values in z-matrix as scan information. 0 1 2 Default (No). Yes. No.IOp(1/61)How ONIOM should leave the rwf at the end of each geomtry: 0 Default (1).1 Normal: leave the rwf set up for the low-level calculation on the real system. 2 MOMM: leave the rwf set up for the real system, but with NBasis and NBsUse for the high-level calc on the model system. Useful for treating the full system as having electrons only on the QM atoms.IOp(1/62)Counterpoise control. NN NN fragments, NN < 50.IOp(1/63)Step in counterpoise calculation: MNN NN = 0 1-NFrag M = order of derivatives (1=Energy, 2=Gradient, Supermolecule Fragments with ghost atomsNFrag+1 - 2*NFrag -- lone fragmentsIOp(1/64)Molecular mechanics force field selection: 0 1 2 3 4 5 6 7 000 100 200 300 None. Dreiding. UFF. AMBER. MM2 (NYI). MM3 (NYI). MMFF (NYI). Quartic fitting field (NYI). Use only hard-wired. Use soft and hard-wired, hard-wired has priority. Use soft and hard-wired, soft has priority. Use only soft. Lowest 2 digits then have no meaning.0000 Do not read modifications to parameter set. 1000 Read modifications to parameter set. 00000 With soft parameters, abort when different parameters match to the same degree. 10000 Use the first when there are equivalent matches. 20000 Use the last when there are equivalent matches. If IOp(67)=3, then the default is to apply soft parameters with higher priority.IOp(1/65)Control of which terms are included in MM, corresponding to the 'classes' in FncInf. 0 1 10 100 1000 10000 Do all (default) Non-bonded Stretching Bending Torsion Out-of-planeIOp(1/66)Whether to generate QEQ charges, over-written the values in AtChMM, or to use the values already there. 0 1 2 00 10 20 000 100 200 Default (2, 1==> 221) Do QEq. Don't do QEq. Default (10) Do for atoms which were not explicitly typed. Do for all atoms regardless of typing. Default (100) Do for atoms which have charge specified or defaulted to 0. Do for all atoms regardless of initial charge.IOp(1/67)Source of MM parameters. 0 1 2 3 Default: 2 if reading geom from chk file, else 1. Generate here, reading from input if requested by IOp(64). Copy from chk file. Pick up non-standard parameters from chk file.IOp(1/70)L118 Type of sampling (Nact) 0 1 2 3 4 Defalt (same as 3) Orthant sampling Microcanonical normal mode sampling Fixed normal mode energy Local mode sampling ( now only Nact = 0 or 3 OK )IOp(1/71)Whether to print out input files for each structure along an IRC: 0 1 No. Yes.IOp(1/72)L103: Algorithm choice for microiterations. L121: Lagrangian constrain method for ADMP (ICType) Half*Gamma*Tr[(P*P-P)**2] + Lambda*[Tr(P)-Ne] + Eta*Tr(P*P-P) 0 Default Same as 7 if no Mass-Weighting (IOp(76) < 0) Same as 10 if Mass-Weighting (IOp(76) > 0) 1 2 Use Lambda and Eta only. (Gamma=0) Use Lambda, Eta, Gamma. Gamma = .23 Use Lambda, Eta, Gamma. Gamma = 1. Constraints for scalar Mass case: 4 Use exact constraint Sum(ij)[Vij*(P**2-P)ij]5-7 Iterative Scheme same as 4. Different initial guesses. 7 is default for scalar mass case. Constraints for tensorial Mass: 8-11 Mass-weighting constraints. Documentation maybe found in DVelV1. 10 is default.IOp(1/73)L103: NInit for microiterations. L121: Initial Kinetic energy of the Nuclei (EStrtC) 0 Default (.1 Hartree)N>0 N*micro-Hartree N<0 0.0 HartreeIOp(1/74)Charge scaling for charge embedding in ONIOM. IJKLMN 6th through 1st nearest neighbors of current layer scaled by I*0.2, J*0.2, etc. 0 ==> 5 (no scaling); all layers are scaled by at least as much as ones farther out. The default is 500. M L0 Factor for charges one bond away from link atom Factor for charges two bonds away from link atomK00 Factor for charges three bonds away from link atom IJ etc. The actual factors used are: 0: 1.0 1: 0.0 2: 0.2 3: 0.4 4: 0.6 5: 0.8 6-9: 1.0IOp(1/75)ADMP control flag (ICntrl) 0 1 2 3 00 10 20 Standard ADMP Read converged density at every step Fix the nuclear coordinates Test time reversability (MaxStp must be even) Default (20). Read stopping parameters from input. Do not read stopping parameters.IOp(1/76)+/- XXXXZYYYY = Ficticous electron mass (EMass) YYYY Default (1000) IOp(76)>0 YYYY*.0001 AMU MW core functions more than valence functions. IOp(76)<0 YYYY*.0001 AMU. Use uniform scaling for all basis functions (Note YYYY > 9999 makes no sense) Z Mass-weighting option. If IOp(76)<0, Z is meaningless.XXXX If PBC: Mass of Box Coordinates (BoxMas) = XXXX*.0001 AMU BoxMas=0 Box coordinates not propagated (default).IOp(1/77)Initial Kinetic energy of the density matrix (EStrtP) (For UHF, Alpha and Beta each get half this energy) and Option Number to compute initial kinetic energy. Format of Input: XXYYYY (six digits) IWType = XX N = YYYY (For UHF, Alpha and Beta each get half this energy) 0 Default (0.0 Hartree)N>0 N*micro-Hartree IWType is used to figure out how the initial velocity is is computed (in gnvelp). If XXYYYY < 0 : Initial velocity = 0.0 Hartee (i.e., currently same as N=0 above)IOp(1/78)Sparse in L121 -N 0 1 Sparse here with cutoff 10**(-N), full elsewhere Use full matrices or spase based on standard settings. Use sparse fixed formIOp(1/79)IRCMax convergence in L115 Stopping criteria in L118 and L121.IOp(1/80)L106: 0/1/2 Cartesian/Normal mode/Internal coordinate differentiation. 2 is NYI. L118: .eq.1 to surpress the 5th order correction after surface hop has been made in Trajectory Surface Hopping calculations. Needs also IOp(10/80=1) Nuclear Kinetic Energy Thermostat Option. (Currently only Velocity scaling is implemented) 0 No Thermostat.11XXXXX Velocity scaling, but only for the first XXXXX simulation steps. (This options is useful, if thermostating in only required during equilibration. 1000000 Velocity scaling, all the way through the simulation.IOp(1/81)Nuclear KE thermostat in ADMP -- temperate is checked and scaled every IOp(81) steps.IOp(1/82)Temperature for nuclear KE thermostat in L121.IOp(1/83)Whether to read in frequencies for electric and magnetic perturbations. 0 1 2 Default (No). Yes. No.IOp(1/84)Differentiation of frequency-dependent properties. 0 N No. Mask for which properties on file 721 will be differentiated.IOp(1/85)Band gap calculation in PBC ADMP: 0 1 2 Default (No). Diagonalizae Fock matrix to get band gap, evolution, etc. No.IOp(1/86)Printing for NMR for ONIOM. 0 1 2 Default (1). Print tensors and eigenvalues. Print eigenvectors as well.IOp(1/87)ONIOM integration of density. 0 1 2 K0 L00 Do not integrate. Integrate current densities. Integrate densities specified by following digits: Density to use from gridpoint 1 Density to use from gridpoint 2M000 etc. K,L,M,etc: 0: SCF 1: MP first order 2: MP2 3: MP3 4: MP4 5: CI one-particle 6: CI 7: QCI/CC 8: Correct to second orderIOp(1/88)Whether to read in atomic masses (isotopes): 0 Default (1 if geometry read from input, 4 if geometry read from chk) 1 Use most abundant isotopes.2 Read isotopes from input. The temperature and pressure are read first, for backwards compatibility. 3 4 Read isotopes from rwf. Read isotopes from chk.IOp(1/89)Maximum allowed deviation from average nuclear KE during ADMP, in Kelvin.IOp(1/90)To read in the velocity in cartesian coordinates Nuclear Kinetic Energy Thermostat Option. Average energy (in microhartree) to be maintained during Simulation, as required by IOp(80).IOp(1/91)Thermostat Option.IOp(1/92)Maximum allowed deviation from average nuclear KE specified in IOp(81). Also in microhartree. IOp(1/94, 95, 96, 97, 98) IOp(94): Davidson control for quadratic micro-iterations (see MMOpt2) IOp(95): RFO/Davidson control for quadratic microiterations (see MMOpt2) IOp(96): Davidson control for coupled QM/MM macro step (see MMOpt2)IOp(97):RFO/Davidson control for coupled QM/MM macro step (see MMOpt2)IOp(98):Control of quadratic micro-iterations and coupled QM/MM quadratic macro step. <0 0 1 2 3 4 5 10 20 30 40 50 Do not use dynamic convergence criteria for the micro-iterations. Default(15). Regular non-coupled macro step. Coupled macro step, full diagonalization. Coupled macro step, direct /w full Hessian incore. Coupled macro step, direct /w MM Hessian incore. Coupled macro step, fully direct. Regular micro-iterations. Quadratic micro-iterations, full diagonalization. Quadratic micro-iterations, direct /w prepared Hessian incore. Quadratic micro-iterations, direct /w raw MM Hessian incore. Quadratic micro-iterations, fully direct.IOp(1/101, 102, 103, 104)Phase control in L115 and L118: N1, N2, N3, N4IOp(1/105)Reaction direction 00 10 Default (Same as 10) Forward direction20 Reverse direction Damped-Velocity Verlet (DVV) options for Dynamic Reaction Path Following 0 1 2 00 10 20 000 100 200 step 300 0000 1000 2000 Default (Same as 2) Use DVV Do not use DVV Default (Same as 10) Follow the rxn path in the forward direction Follow the rxn path in the reverse direction Default (Same as 200) Time step correction not used Time step correction used but not to recalculate current DVVTime step correction used and current DVV step recalculated Default (Same as 1000) Use DVV stopping criteria Do NOT use DVV stopping criteriaIOp(1/106)Damping constant for DVV Dynamic Rxn Path following (v0) 0 N Default v0=0.04 (N=400) v0 is set to N*0.0001IOp(1/107)Error tolerance for DVV time step correction (Error) 0 N Default Error=0.003 (N=30) Error=N*0.0001IOp(1/108)Gradient magnitude for DVV stopping criteria (Crit1) 0 N Default (N=15) N*0.0001IOp(1/109)Force-Velocity angle for DVV stopping criteria (Crit2) 0 N Default (90 Degrees) Use N DegreesIOp(1/110)Scaling of rigid fragment steps during microiterations. 0 1 2 -n Do not scale Scale with 1/NRA (NRA = number of atoms in fragment)Scale with 1/Sqrt(NRA) Scale with 1/nIOp(1/111)Step-size to use with steepest descent when L103 is having trouble: -N -1 0 N Scale up to RMS step of N/1000 if DXRMS is less. Effectively disables the scaling Default (50) Scale up or down to maximum change in a variable of N/1000IOp(1/112)Temperature for thermochemistry. 0 N Default (standard temperature, unless read in). N/1000 degrees.IOp(1/113)Pressure for thermochemistry. 0 N Default (1 atomosphere, unless read in). N/1000 atmospheres.IOp(1/114)Scale factor for harmonic frequencies for use in thermochemistry and harmonic vibration-rotation analysis. 0 N Default (1 unless specified by IOp in overlay 7 or read in). N/1000000.。
Gaussian简介Gaussian简介Gaussian是做半经验计算和从头计算使用最广泛的量子化学软件,可以研究:分子能量和结构,过渡态的能量和结构化学键以及反应能量,分子轨道,偶极矩和多极矩,原子电荷和电势,振动频率,红外和拉曼光谱,NMR,极化率和超极化率,热力学性质,反应路径。
计算可以模拟在气相和溶液中的体系,模拟基态和激发态。
Gaussian 03还可以对周期边界体系进行计算。
Gaussian是研究诸如取代效应,反应机理,势能面和激发态能量的有力工具。
功能①基本算法②能量③分子特性④溶剂模型Gaussian03新增加的内容①新的量子化学方法②新的分子特性③新增加的基本算法④新增功能(1)基本算法可对任何一般的收缩gaussian函数进行单电子和双电子积分。
这些基函数可以是笛卡尔高斯函数或纯角动量函数多种基组存储于程序中,通过名称调用。
积分可储存在内存,外接存储器上,或用到时重新计算对于某些类型的计算,计算的花费可以使用快速多极方法(FMM)和稀疏矩阵技术线性化。
将原子轨(AO)积分转换成分子轨道基的计算,可用的方法有in-core(将AO积分全部存在内存里),直接(不需储存积分),半直接(储存部分积分),和传统方法(所有AO 积分储存在硬盘上)。
(2)能量使用AMBER,DREIDING和UFF力场的分子力学计算。
使用CNDO, INDO, MINDO/3, MNDO, AM1,和PM3模型哈密顿量的半经验方法计算。
使用闭壳层(RHF),自旋非限制开壳层(UHF),自旋限制开壳层(ROHF) Hartree-Fock 波函数的自洽场SCF)计算。
使用二级,三级,四级和五级Moller-Plesset微扰理论计算相关能。
MP2计算可用直接和半直接方法,有效地使用可用的内存和硬盘空间用组态相互作用(CI)计算相关能,使用全部双激发(CID)或全部单激发和双激发(CISD)。
双取代的耦合簇理论(CCD),单双取代耦合簇理论(CCSD),单双取代的二次组态相互作用(QCISD), 和Brueckner Doubles理论。
Gaussian关键字Chapter 2 单点能计算(Single Point Energy Calculations)The Route Section#T 只输出重要的结果(terse output)#正常Gaussian输出(traditional)#P详细输出Test任务结果不写入site archivePop=Reg只显示5个最高占据轨道和5个最低未占轨道(Molecular Orbital Coefficients)Pop=Full显示全部轨道(Molecular Orbital Coefficients)Units说明在molecule specification中使用不同的单位SCF=Tight要求波函数更严格地收敛多步任务(Multi-step Jobs)在两个任务之间加“--Link1--”语句,且“--Link1--”之前要有一空行,即:…………--Link1--…………--Link1--…………计算NMR shielding tensors: NMRe.g. #T RHF/6-31G(d) NMR Test--------------------------------------------------------------------------------------------------------------------------------- e.g. %Chk=benzene#T B3LYP/6-31G(d) Opt Test……(title; molecule specification)--Link1—%Chk=benzene%NoSave#T HF/6-311+G(2d,p) NMR Geom=AllCheck Guess=Read Test测试波函数的稳定性:Stable 包括确定对于目标,是否存在能量更低的波函数,通过释放默认的约束来实现(例如让波函数变为开壳层或降低轨道的对称性)Stable=opt当存在不稳定性时,重新优化波函数(与几何优化不同)e.g. #T RHF/6-31G(d) Stable Test也可以加入Guess=Mix或Guess=Mix Stable命令chapter 3 结构优化(Geometry Optimizations)寻找极小点:通过搜索“converged?”字符串确定优化是否完成STQN方法:QST2e.g. #T UHF/6-31G(d) Opt=QST2 TestH3CO --> H2COH Reactants0 2structure for H3COH3CO --> H2COH Products0 2structure for H2COH力常数的计算:Opt=ReadFC从事先已有的作频率计算的check文件(通常用较低的方法或较小的基组)中读取力常数,而不是估计(estimate)。
1 计算流程上面的图应该是从L301说起的,即图中的“基组”,L301的作用是产生基组信息。
(在这之前还有L1:处理计算执行路径,创建执行链接的列表,并初始化scratch文件;L101:读取标题和分子说明部分;L103:berny优化到最小值;L202:重新定位坐标,计算对称性,检查变量)“基组”之后是“分子结构”即L401,形成初始的分子轨道初猜。
第一个框中有“分子轨道”和“能量”两项,我想这应该指的是L502即迭代求解SCF方程。
下面红线圈的框中则是迭代求解的具体过程。
每次迭代完毕都得到一个能量。
即(SCF Done: E(RHF) = -xx.xxxx A.U. after xx cycles)然后是L103判断力和位移是否满足收敛条件。
如果满足(即四个yes),则进行布居分析等,并完成计算。
如果不满足,则继续调整分子结构(L401),再次进行迭代求解,反复这个过程,直到满足力和位移收敛条件为止。
如果不是HF方法而是多体微扰,CI等后-scf方法的话,在每次迭代(L502)完成后还要多一步相关能的计算,以MP2为例,迭代完成后多出:L801(双电子积分变换的初始化),L906(半直接的MP2)和L1002(迭代求解CPHF方程)等计算。
对于楼主的图,我觉得力和位移判断为NO以后,箭头应该指向最头上的“基组”,因为实际过程中,每次构型调整都会重新定位坐标和产生基组信息。
常见问题分析1.检查是否有初始文件错误在命令行中加入 %kJob L301 or %kJob L302 如果通过则一般初始文件ok。
常见初级错误:a. 自旋多重度错误; b. 变量赋值为整数; c. 变量没有赋值或多重赋值; d. 键角小于等于0度,大于等于180度e. 分子描述后面没有空行; f. 二面角判断错误,造成两个原子距离过近; g. 分子描述一行内两次参考同一原子,或参考原子共线2. SCF(自洽场)不收敛则一般是L502错误,省却情况做64个cycle迭代(G03缺省128 cycles)a. 修改坐标,使之合理;b. 改变初始猜 Guess=Huckel 或其他的,看Guess关键词;c. 增加叠代次数 SCFCYC=N (对小分子作计算时最好不要增加,很可能结构不合理); d. iop(5/13=1)这样忽略不收敛,继续往下做。