用Gaussian 和GaussView 计算分子的频率
- 格式:pdf
- 大小:2.60 MB
- 文档页数:8


Gaussian和GaussView在结构化学教学中的应用黄钦【摘要】Structural Chemistry was a theoretical strong curriculum.The application of Gaussian and GaussView in the Structural Chemistry curriculum teaching process was discussed via the example of molecular structure of cyclopentadiene and cyclopentadiene anion,molecular orbital of hydrogen fluoride,and IR principle of water molecule.This method was conducive to breakthrough in teaching difficulties,and to improve teaching quality.%结构化学是一门理论性较强的基础课程。
本文结合环戊二烯分子和环戊二烯负离子的分子构型,HF分子的分子轨道和H2O分子的红外光谱原理等实例,对Gaussian和GaussView在结构化学课程教学过程中的应用进行了有意义的探讨,这种形象的教学方法有利于突破教学难点,提高教学质量。
【期刊名称】《广州化工》【年(卷),期】2012(040)010【总页数】3页(P199-200,202)【关键词】结构化学;Gaussian;GaussView【作者】黄钦【作者单位】广西民族大学化学化工学院,广西南宁530006【正文语种】中文【中图分类】O641物质由分子构成,分子的结构决定了物质的性质。
结构化学是在原子、分子的水平上深入到电子层次,研究物质的微观结构及其与宏观性质间的关系的学科,对于使学生建立初步的微观结构原理,培养科学精神和创新思维,具有不可替代的作用[1]。

优化1在优化时采用Scf(tight)的选项,增加收敛的标准。
再去计算频率。
如果还有虚频,参见下一步。
2.对称性的影响,很多情况下的虚频是由于分子本身的对称性造成的。
这样,在优化时,如果必要,要将对称性降低,还有,输入文件有时是用内坐标。
建议如果有虚频的话,将内坐标改成直角坐标优化。
3.如果上述方法还有虚频,看一下虚频,找到强度较大的,将在频率中产生的原子的振动坐标加到相应的输入文件中。
这样,重新计算。
直到虚频没有。
4实际上,如果分子柔性较大,很难找到最低点,这是电子结构计算的问题,这种情况下,需要动力学的东西,用构象搜寻的办法解决。
如:模拟退火,最陡下降法,淬火法等。
将得到的能量最低的构象做一般的电子结构计算,这样,应当没有问题。
不要讲你还没有得到最稳定的结构,那么,是你的分子有问题,要么计算错了,要么就是游离出现代计算的范畴1。
低频率振动模式时,力常数很小,必须使用opt=tight以确保适当的收敛和随后任务步骤中频率计算的可靠度。
比如我曾经算过一个体系,第一个频率振动的力常数只有10~20,很不放心,加入tight后,优出来的结构却与前面大相径庭,而且还出现了一个虚频。
2。
做IRC计算的时候,如果步长很小,必须要用verytight。
就曾有人步长设为1,而不加verytight,算出来的反应路径是v字形倒海鸥状的。
3。
使用int指定用于数值积分的积分网格来用于消虚频的时候,必须注意,在比较能量的时候,对所有的计算要使用相同的积分网格。
检查错误1.检查是否有初始文件错误在命令行中加入%kJob L301 or %kJob L302如果通过则一般初始文件ok。
常见初级错误:a.自旋多重度错误b.变量赋值为整数c.变量没有赋值或多重赋值d.键角小于等于0度,大于等于180度e.分子描述后面没有空行f.二面角判断错误,造成两个原子距离过近g.分子描述一行内两次参考同一原子,或参考原子共线2.SCF(自洽场)不收敛则一般是L502错误省却情况做64个cycle迭代(G03缺省128 cycles)a.修改坐标,使之合理b.改变初始猜Guess=Huckel或其他的,看Guess关键词。

gaussian频率计算Gaussian频率计算是一种常用的计算化学方法,用于研究分子的振动和光谱性质。
在化学和材料科学领域中,频率计算是理解和解释分子结构、反应机理和光谱谱线的重要工具。
本文将详细介绍Gaussian频率计算的原理和应用,并探讨其在科学研究中的重要性。
我们来了解一下Gaussian频率计算的基本原理。
频率计算是基于量子力学的原理,通过求解分子的力常数矩阵来获得分子的振动频率。
在Gaussian软件中,通过输入分子的几何构型和相关参数,利用量子力学的原子核运动方程求解,得到分子的振动频率和振动模式。
这些振动频率可以用来计算分子的红外光谱、拉曼光谱以及其他光谱性质,从而进一步研究分子的结构和性质。
Gaussian频率计算的应用非常广泛。
首先,它可以用于确定分子的构型和几何参数。
通过计算不同构型下的振动频率,可以确定分子的平衡几何构型和键长、键角等几何参数。
这对于理解分子的稳定性、反应机理以及与其他分子的相互作用非常重要。
Gaussian频率计算可以用于预测和解释分子的光谱性质。
振动频率与分子的红外光谱和拉曼光谱密切相关。
通过计算分子的振动频率和振动模式,可以预测和解释实验观测到的红外光谱和拉曼光谱谱线。
这对于确定分子的化学键、官能团和分子结构非常有帮助。
Gaussian频率计算还可以用于研究分子的振动性质和能量。
通过计算不同振动模式的振动能量和振动强度,可以了解分子的振动能级和振动强度分布,从而进一步研究分子的能量分布和振动态。
Gaussian频率计算在材料科学中也有重要应用。
振动频率对于材料的力学性质、热学性质和光学性质具有重要影响。
通过计算材料的振动频率和振动模式,可以研究材料的弹性、热膨胀、热导率、光学吸收等性质,为材料设计和性能优化提供指导。
Gaussian频率计算是一种重要的计算化学方法,可以用于研究分子的振动和光谱性质。
它在理论研究、实验解释和材料设计等方面都具有广泛的应用。


Course Education Research2018年第33期课程教育研究科学·自然Gaussian09和GaussView软件在结构化学教学中的应用戴国梁(通讯作者)钱蕙(苏州科技大学化学生物和材料工程学院江苏苏州215009)【摘要】作为化学专业的基础课程,结构化学由于其抽象性及理论性强等特点,给教和学双方均带来了一定困难。
本文作者结合自己的专业研究方向,在结构化学讲授中应用Gaussian09和GaussView等软件对分子轨道等相关知识点进行辅助教学,使抽象的化学理论变得形象、简单,这对培养学生的形象思维能力和学习兴趣,提高本课程的教学效果有很大的帮助。
【关键词】Gaussian软件辅助教学结构化学教学方法【基金项目】江苏省高校自然科学基金,项目编号:14KJB150024。
【中图分类号】G64【文献标识码】A【文章编号】2095-3089(2018)33-0156-011.课程特点和软件简介当今化学已进入纳米空间、皮秒时间时代,随着人们对物质微观结构认识的不断深入,结构化学的基本理论越来越广泛地应用于化学的各个领域,特别是在材料、信息、能源等领域。
结构化学是研究原子、分子、晶体的结构以及结构与性质之间关系的科学。
相比化学专业的其他基础课程,本课程的新概念多,数学推导多,系统性很强,故而要求学生在学习过程中既有严密的逻辑思维能力,还要有较好的空间想象力,特别是要具备有一定的数学、物理基础,才能获得较好的教学效果。
与其他化学基础课程如有机,无机,分析化学等不同,本课程的学习过程中缺少相关的实验演示和操作,而其他化学类课程在理论学习的同时,均开设同步的实验课程。
本课程的学习需要较多的数学、物理知识,而很多师范院校化学专业学生数理基础相对薄弱,教学过程中不得不给学生补充一些线性代数知识,造成实际讲授该课程的学时数偏少。
同时,学生在学习过程中往往对一些繁琐的数学公式及其推导过程有厌倦和抵触心理,故学生对结构化学学习的积极性不高。

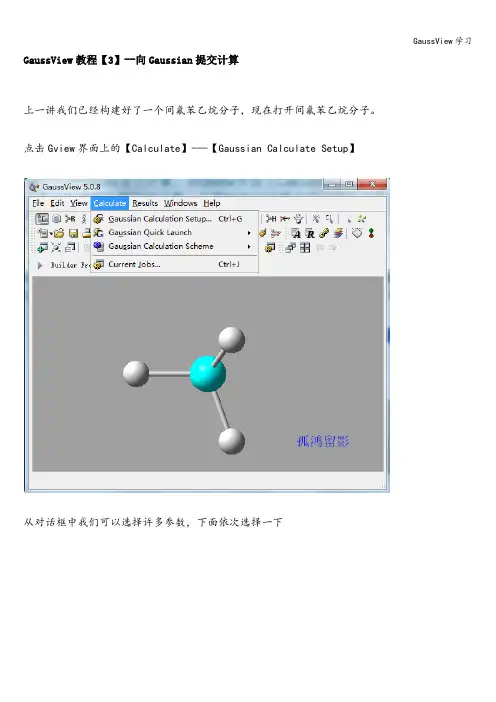
GaussView教程【3】--向Gaussian提交计算上一讲我们已经构建好了一个间氟苯乙烷分子,现在打开间氟苯乙烷分子。
点击Gview界面上的【Calculate】-—【Gaussian Calculate Setup】从对话框中我们可以选择许多参数,下面依次选择一下【Job Type】工作类型:【Energy能量】|【Optimization优化】|【Frequency频率】|【Opt+Freq优化+频率】|【Scan扫描】|【Stability稳定性】|【NMR核磁】,这里我们选择【NMR】核磁【Method】方法:每种计算模式都提供了若干种方法,这里选择默认值即可【Title】题目:这项可以根据自己的项目自行命名【Link 0】给检查点文件命名其它选择默认即可选好后点击【Submit】提交至Gaussian,并保存。
(最好存到安装文件所在磁盘)系统会询问是否提交至Gaussian,选择【OK】,Gaussian会自动开启并计算,计算时间因硬件配置而异计算完毕后系统会提示关闭Gaussian,点击【是】计算完毕后会生成两个文件,.log的是系统日志,便于查看计算结果。
这里我们选择.chk文件【Ok】接下来我们又一次看到了刚才绘制的那个分子,表面上没什么不同,但这次我们可以预言它的许多性质了,在GView的主界面点击【Results】-—【Summary】可以看到刚才的计算总结,点击【View File】可以看到日志文件。
因为我们刚才选择计算模式是【NMR】核磁,所以我们点击【Results】——【NMR】,随即我们看到了很熟悉的核磁图谱,但似乎很乱,因为这是所有元素的核磁谱图,在左下方【Element】右方选择【H】,即H谱,旁边【Reference】选择【TMS HF/6—31G(d)】,这次我们就可以看到间氟苯乙烷分子理论计算的核磁谱图了。
在绘图界面右击【View】——【Labels】可以看到每个H原子对应场中的位置。

Gaussian 程序频率计算中的几个问题在Gaussian计算中,为了确定优化得到的几何结构是势能面上的局域极小点还是鞍点,或者要得到相关的热力学性质,经常需要对优化后的几何结构进行振动分析。
这里我们将讨论几个频率计算中常见的一些问题。
希望能对初学Gaussian的人有所帮助。
首先,原则上说,振动频率分析只对稳定结构有意义。
这里所说的稳定结构包括是势能面上的局域极小点和鞍点。
如下图1所示是一维自由度上的势能面,A和B处在势能面的局域极小点,而处在势能面的鞍点上。
他们在都处在平衡位置(原子核受力为零),不同的是,A和B来说离开平衡位置会受到指向平衡位置处的力,而C离开平衡位置会受到远离平衡位置的力。
因此A和B处在稳定平衡点,C处在不稳定平衡点。
实际上,一个分子可以有很多的自由度,如果在所有自由度上分子都处在稳定平衡,就是稳定的分子。
频率分析得结果是所有频率都是正的,表明这是一个局域的极小点。
如果分子只在一个自由度上处于不稳定平衡位置,其他自由度上都处在稳定平衡位置,说明该结构是一阶鞍点。
分子在稳定自由度方向上的振动才是真实的振动,在不稳定自由度方向上的实际上是不会有振动的。
不过我们可以对不稳定方向上的运动也按振动来做数学处理,会的到负的振动频率,我们称它为虚频。
虚频的出现表明该结构为鞍点。
图1 势能面上的局域极小点和鞍点第二,Gaussian计算中,频率的计算一定要在和分子结构优化相同的方法,基组下进行,否则计算的结果是没有意义的。
我们知道,任何理论水平下的计算,都是在一定的近似下进行的,不同的理论水平的近似程度是不同的。
在一种理论水平A下优化的稳定结构Geom_A会和另一种理论水平B下优化的稳定结构Geom_B有差别,也就是说Geom_A不会是理论水平B下的稳定结构。
根据前面我们所讨论的,在理论水平B下对一个不稳定的结构进行频率分析是没有意义的。
图2示意说明了不同理论水平下稳定点结构的不同。
图2 不同理论水平下优化的稳定结构是不同的第三,频率计算中可以考虑同位素效应(Freq=ReadIsotopes)。

gaussian 09w计算实例
Gaussian 09W是一种用于计算化学结构和性质的量子化学软件,它可以进行从简单的分子力场计算到复杂的量子力学计算等多种类
型的计算。
下面我将从几个方面介绍Gaussian 09W的计算实例。
首先,Gaussian 09W可以用于分子结构优化。
通过输入分子的
初始几何结构和所需的计算方法,Gaussian 09W可以利用量子力学
方法对分子的结构进行优化,找到最稳定的构型,并给出最低能量
的构型。
其次,Gaussian 09W可以用于计算分子的振动频率和红外光谱。
通过输入优化后的分子结构,Gaussian 09W可以计算分子的振动频
率和红外光谱,从而帮助研究者理解分子内部原子的振动特性和分
子与外界光的相互作用。
此外,Gaussian 09W还可以用于计算分子的电子结构和性质。
通过输入分子的结构信息和所需的计算方法,Gaussian 09W可以计
算分子的电子结构、电离能、电子亲和能等性质,帮助研究者深入
了解分子的电子行为和化学性质。
另外,Gaussian 09W也可以用于计算分子间相互作用和反应动力学。
通过输入反应物和所需的计算方法,Gaussian 09W可以模拟分子间的相互作用和化学反应过程,帮助研究者理解和预测化学反应的动力学行为。
综上所述,Gaussian 09W是一款功能强大的量子化学软件,可以广泛应用于分子结构优化、振动频率和红外光谱计算、电子结构和性质计算以及分子间相互作用和反应动力学等多个方面的计算。
通过合理选择输入参数和方法,研究者可以利用Gaussian 09W进行全面而深入的分子计算研究。

•理论计算研究通常的流程(性质和反应机理)选择一定的理论和基组对结构进行优化,得到基态分子的各种性质可以用TD理论进行激发态理论研究预测紫外光谱、荧光光谱等用相同的理论和基组来寻找过渡态的优化结构,具有一个虚频IRC分析确保找到的过渡态正确连接了原料和产物。
1. 计算方法和模型化学计算化学的方法主要有1. 分子力学理论(Molecular Mechanics)•分子力学理论采用经典物理对分子进行处理,可以在MM3,HyperChem,Quanta,Sybyl等软件中使用。
根据所采用的力场的不同,分子理论又分为很多种。
2. 电子结构理论(ElectronicStructure Theory)•这一理论基于薛定鄂方程,采用量子化学方法对分子进行处理•1). 半经验的方法•2). 从头计算2. 单点能的计算单点能计算是指对给定几何构性的分子的能量以及性质进行计算,由于分子的几何构型是固定不变的,只是"一个点",所以叫单点能计算。
•可以作为分子构型优化前对分子的检查•在由较低等级计算得到的优化结果上进行高精度的计算3. 分子结构的优化寻找极小值•有一个初始结构•对初始结构进行改变,寻找能量更小的结构•当达到一定的收敛标准时•得到稳定点4. 频率分析频率分析可以用于多种目的:•预测分子的红外和拉曼光谱(频率和强度)•为几何优化计算力矩阵•判断分子在势能面上的位置•计算零点能和热力学数据如系统的熵和焓环状亚胺的频率分析4. 化学反应机理的计算马鞍面+反应原料反应产物过渡态4. 化学反应机理的计算过渡态+5. 分子的激发态性质Gaussian可以预测紫外光谱、荧光光谱等、并可以计算激发态的结构和性质,计算光化学反应机理等。
苯转变成盆烯的光化学反应[*]光解6. 化学反应中的溶剂效应很多有机化学反应是在有机溶剂中开展的,而通常的Gaussian理论计算只是在真空条件下的计算。
Gaussian建立了很多种溶剂的模型,可以将有机分子置于有机溶剂环境中进行结构优化和反应机理模拟。
Gaussian安装一、先安装Gaussian09(手动选择安装在D盘Gaussian文件夹,序列号在pin文件中,其他依次next即可);二、再安装Gaussian VIEW05(输入文件与输出文件类型全选)。
Gaussian操作一、打开GaussView,同时出现两个窗口GaussView 5.0.9与G1:M1:V1-New。
二、构建分子模型:在如下图中GaussView 5.0.9窗口中点击红色椭圆圈住的地方,如果构建水分子直接点元素周期表O原子;如果是构建葡萄糖分子就需要先在GaussView 5.0.9窗口红色椭圆圈住的地方点C原子,然后在G1:M1:V1-New窗口空白处左击鼠标一下会出现一个CH4分子,接着在GaussView 5.0.9窗口红色椭圆圈住的地方点O原子,再在G1:M1:V1-New窗口中点击刚才出现的CH4分子上的一个氢原子,同样道理依次进行下去便可构建一个单葡萄糖分子,从而用来模拟。
三、鼠标右击GaussView 5.0.9中建好的分子界面,选择Calculate——Gaussian Calculation Setup进入Gaussian Calculation Setup设置界面。
四、设置如下几项:1.Job Type :Opt + Freq(优化加频率,其他不变)2.Method(如下设置都是参考北师大计算glucose)Method(方法):Ground State,DFT, Restricted, B3LYPBasis Set(基组):6-31G, +,d3.完成后点下方“Submit(提交)”,弹出如下对话框,选择Save,可以自主选择保存到相应文件夹并自行命名(注意:文件夹名与文件名必须为英文或数字,不可有汉字,否则计算会出现问题)。
4.确定文件夹及文件名后,点击Save会弹出如下对话框,选择OK,电脑会自行链接打开Gaussian09开始计算,计算结束会出现完成报告(水分子大概7s时间),选择是。
gauc计算Gaussian计算是一种常用的计算化学方法,它可以用来计算分子的结构、能量、振动频率、光谱等性质。
Gaussian是一款商业软件,由Gaussian Inc.开发,目前已经发展到了Gaussian 16版本。
本文将介绍Gaussian计算的基本原理、使用方法以及一些常见的应用。
一、Gaussian计算的基本原理Gaussian计算的基本原理是量子力学。
在量子力学中,分子的运动状态可以用波函数来描述。
Gaussian计算就是通过求解分子的波函数来计算分子的性质。
具体来说,Gaussian计算可以分为以下几个步骤:1.构建分子模型。
在Gaussian中,可以通过手动输入分子的坐标或者从文件中读取分子的坐标来构建分子模型。
2.选择计算方法。
Gaussian支持多种计算方法,包括Hartree-Fock方法、密度泛函理论方法、半经验方法等。
不同的计算方法适用于不同的分子体系和性质计算。
3.设置计算参数。
在进行计算之前,需要设置一些计算参数,如基组、收敛标准、计算级别等。
这些参数的选择会影响计算结果的精度和计算时间。
4.进行计算。
在设置好计算参数之后,就可以进行计算了。
Gaussian会自动求解分子的波函数,并计算分子的性质。
5.分析计算结果。
计算完成后,需要对计算结果进行分析。
Gaussian可以输出分子的结构、能量、振动频率、光谱等性质,可以通过这些性质来判断计算结果的可靠性和分子的性质。
二、Gaussian计算的使用方法Gaussian计算是一款非常强大的计算化学软件,但是对于初学者来说,使用起来可能会比较困难。
下面介绍一些Gaussian计算的使用方法,帮助初学者更好地使用这款软件。
1.学习基本原理。
在使用Gaussian之前,需要先学习一些基本的量子化学原理,如分子轨道理论、Hartree-Fock方法、密度泛函理论等。
只有掌握了这些基本原理,才能更好地理解Gaussian计算的原理和方法。
在Gaussian计算中,为了确定优化得到的几何结构是势能面上的局域极小点还是鞍点,或者要得到相关的热力学性质,经常需要对优化后的几何结构进行振动分析。
这里我们将讨论几个频率计算中常见的一些问题。
希望能对初学Gaussian的人有所帮助。
首先,原则上说,振动频率分析只对稳定结构有意义。
这里所说的稳定结构包括是势能面上的局域极小点和鞍点。
如下图1所示是一维自由度上的势能面,A和B处在势能面的局域极小点,而处在势能面的鞍点上。
他们在都处在平衡位置(原子核受力为零),不同的是,A和B来说离开平衡位置会受到指向平衡位置处的力,而C离开平衡位置会受到远离平衡位置的力。
因此A和B处在稳定平衡点,C处在不稳定平衡点。
实际上,一个分子可以有很多的自由度,如果在所有自由度上分子都处在稳定平衡,就是稳定的分子。
频率分析得结果是所有频率都是正的,表明这是一个局域的极小点。
如果分子只在一个自由度上处于不稳定平衡位置,其他自由度上都处在稳定平衡位置,说明该结构是一阶鞍点。
分子在稳定自由度方向上的振动才是真实的振动,在不稳定自由度方向上的实际上是不会有振动的。
不过我们可以对不稳定方向上的运动也按振动来做数学处理,会的到负的振动频率,我们称它为虚频。
虚频的出现表明该结构为鞍点。
第二,Gaussian计算中,频率的计算一定要在和分子结构优化相同的方法,基组下进行,否则计算的结果是没有意义的。
我们知道,任何理论水平下的计算,都是在一定的近似下进行的,不同的理论水平的近似程度是不同的。
在一种理论水平A下优化的稳定结构Geom_A会和另一种理论水平B下优化的稳定结构Geom_B有差别,也就是说Geom_A不会是理论水平B下的稳定结构。
根据前面我们所讨论的,在理论水平B下对一个不稳定的结构进行频率分析是没有意义的。
图2示意说明了不同理论水平下稳定点结构的不同。
图2 不同理论水平下优化的稳定结构是不同的第三,频率计算中可以考虑同位素效应(Freq=ReadIsotopes)。
培训课件Gaussian软件的使用频率分析维里定理计算模型化学模型(一)培训课件Gaussian软件的使用频率分析维里定理计算模型化学模型随着科学技术的不断发展,计算化学已成为化学研究中不可或缺的重要分支。
而使用Gaussian计算软件进行频率分析和维里定理计算则是计算化学领域中常见的方法。
在实际工作中,因为涉及到Gaussian的使用,需要进行培训和学习,学习课件就成为了必要的工具。
一、Gaussian软件的使用频率分析Gaussian是一款常用的量子化学软件,它可以用来模拟分子的结构和反应,进行电子结构计算、分子动力学模拟等。
其中,频率分析是Gaussian软件中的一个重要功能。
通过频率分析,可以得到分子的振动信息,包括转动、弯曲、伸缩等各种振动模式,从而获得分子的结构、性质和反应等方面的信息,对化学和材料科学研究具有重要的意义。
二、维里定理计算模型在物理学中,维里定理描述了当物体振动时,其储存的能量和能量损失速率的关系。
在计算化学中,维里定理也有应用。
当分子中原子之间存在相互作用时,若对其进行微小扰动,其振动频率会变化。
因此,可以利用维里定理来计算分子振动频率的变化。
三、化学模型在实际的应用中,通过建立化学模型并使用Gaussian软件进行分析、计算等操作可以获得更加实用和准确的结果。
化学模型可能包括分子结构、反应机理,以及各种化学物性等信息。
基于化学模型,可以构建相应的模型进行计算,从而得到分子结构和反应过程的详细信息。
总之,Gaussian软件的使用频率分析、维里定理计算以及化学模型构建是计算化学领域中非常重要的工具和方法。
通过培训课件的学习,可以掌握Gaussian软件的使用技巧和方法,构建相应的计算模型以及分析计算结果。
这对于化学和材料科学的发展和研究具有重要的意义,也为我们提供了更多的研究手段。
Gaussian View使用手册一、引言Gaussian View是一款用于分子建模和计算化学领域的软件,它提供了一系列强大的功能来帮助用户进行分子结构的建模、分析和计算。
本文将为您介绍如何使用Gaussian View软件进行分子建模和计算的基本操作,包括软件的安装、界面的介绍、分子结构的建立和优化、能量和频率的计算等方面的内容。
二、软件安装与启动1. 下载Gaussian View软件安装包,并按照安装向导逐步进行安装。
2. 安装完成后,双击桌面上的Gaussian View图标,打开软件。
三、界面介绍1. 主窗口:软件的主要操作界面,包括菜单栏、工具栏、分子编辑区、控制台等组件。
2. 分子编辑区:用户可以在此处进行分子结构的建立、编辑和优化操作。
3. 控制台:显示软件运行的状态和输出信息。
四、分子结构的建立与编辑1. 新建分子:点击菜单栏中的“File”-“New”选项,选择分子的基本信息,并在分子编辑区中绘制原子和键。
2. 分子编辑:在分子编辑区中可以通过拖动、旋转、缩放等操作改变分子的结构。
3. 分子优化:点击菜单栏中的“Calculate”-“Optimization”选项,对分子结构进行优化。
五、能量和频率的计算1. 能量计算:点击菜单栏中的“Calculate”-“Single Point Energy”选项,对已优化的分子结构进行能量的计算。
2. 频率计算:点击菜单栏中的“Calculate”-“Frequency”选项,对已优化的分子结构进行振动频率的计算。
六、结果分析与导出1. 结果可视化:在控制台中查看计算过程的输出信息和结果,包括能量、振动频率等数据。
2. 结果导出:将分子结构、能量、振动频率等结果导出为文件,方便后续的分析和报告撰写。
七、常见问题与解决方法1. 软件安装问题:如果在安装过程中遇到问题,可以参考官方的安装文档或联系技术支持。
2. 分子结构优化失败:可能是由于初始结构不合理或参数设置不当,可以尝试调整参数或重新建立分子结构。