水 分 测 定 法(中国药典2010第一部)
- 格式:doc
- 大小:41.00 KB
- 文档页数:4

附录ⅨH. 水分测定法测定用的供试品,一般先破碎成直径不超过3mm的颗粒或碎片。
直径和长度在3mm以下的可不破碎。
减压干燥法需通过二号筛。
第一法(烘干法) 本法适用于不含或少含挥发性成分的药品。
测定法取供试品2~5g,平铺于干燥至恒重的扁形称瓶中,厚度不超过5mm,疏松供试品不超过10m m,精密称定,打开瓶盖在100~105℃干燥5小时,将瓶盖盖好,移置干燥器中,冷却30分钟,精密称定,再在上述温度干燥1小时,冷却,称重,至连续两次称重的差异不超过5mg为止。
根据减失的重量,计算供试品中含水量(%)。
第二法(甲苯法) 本法适用于含挥发性成分的药品。
仪器装置如图。
A为500ml的短颈圆底烧瓶;B为水分测定管;C为直形冷凝管,外管长40cm。
使用前,全部仪器应清洁,并置烘箱中烘干。
测定法取供试品适量(约相当于含水量1~4ml),精密称定,置A瓶中,加甲苯约200ml,必要时加入干燥、洁净的沸石或玻璃珠数粒,将仪器各部分连接,自冷凝管顶端加入甲苯,至充满B管的狭细部分。
将A瓶置电热套中或用其他适宜方法缓缓加热,待甲苯开始沸腾时,调节温度,使每秒钟馏出2滴。
待水分完全馏出,即测定管刻度部分的水量不再增加时,将冷凝管内部先用甲苯冲洗,再用饱蘸甲苯的长刷或其他适宜的方法,将管壁上附着的甲苯推下,继续蒸馏5分钟,放冷至室温,拆卸装置,如有水黏附在B管的管壁放置,使水分与甲苯完全分离(可加亚甲蓝粉末少量,使水染上,可用蘸甲苯的铜丝推下,成蓝色,以便分离观察)。
检读水量,并计算供试品中的含水量(%)。
【附注】用化学纯甲苯直接测定,必要时甲苯可先加水少量,充分振摇后放置,将水层分离弃去,经蒸馏后使用。
第三法(减压干燥法)本法适用于含有挥发性成分的贵重药品。

中国药典2010 年版一部附录附录Ⅰ A 丸剂丸剂系指饮片细粉或提取物加适宜的黏合剂或其他辅料制成的球形或类球形制剂,分为蜜丸、水蜜丸、水丸、糊丸、蜡丸和浓缩丸等类型。
蜜丸系指饮片细粉以蜂蜜为黏合剂制成的丸剂。
其中每丸重量在0.5g(含0.5g)以上的称大蜜丸,每丸重量在0.5g 以下的称小蜜丸。
水蜜丸系指饮片细粉以蜂蜜和水为黏合剂制成的丸剂。
水丸系指饮片细粉以水(或根据制法用黄酒、醋、稀药汁、糖液等)为黏合剂制成的丸剂。
糊丸系指饮片细粉以米粉、米糊或面糊等为黏合剂制成的丸剂。
蜡丸系指饮片细粉以蜂蜡为黏合剂制成的丸剂。
浓缩丸系指饮片或部分饮片提取浓缩后,与适宜的辅料或其余饮片细粉,以水、蜂蜜或蜂蜜和水为勤合剂制成的丸剂。
根据所用黏合剂的不同,分为浓缩水丸、浓缩蜜丸和浓缩水蜜丸。
丸剂在生产与贮藏期间应符合下列有关规定。
一、除另有规定外,供制丸剂用的药粉应为细粉或最细粉。
二、蜜丸所用蜂蜜须经炼制后使用,按炼蜜程度分为嫩蜜、中蜜和老蜜,制备蜜丸时可根据品种、气像等具体情况选用。
除另有规定外,用塑制法制备蜜丸时,炼蜜应雄热加入药粉中,混合均匀;处方中有树脂类、胶类及含挥发性成分的药味时,炼蜜应在60℃左右加入;用泛制法制备水蜜丸时,炼蜜应用沸水稀释后使用。
三、浓缩丸所用提取物应按制法规定,采用一定的方法提取浓缩制成。
四、除另有规定外,水蜜丸、水丸、浓缩水蜜丸和浓缩水丸均应在80℃以下干燥;含挥发性成分或淀粉较多的丸剂(包括糊丸)应在60℃以下干燥;不宜加热干燥的应采用其他适宜的方法干燥。
五、制备蜡丸所用的蜂蜡应符合本版药典该饮片项下的规定。
制备时,将蜂蜡加热熔化,待冷却至60℃左右按比例加入药粉,棍合均匀,趁热按塑制法制丸,并注意保温。
六、凡需包衣和打光的丸剂,应使用各品种制法项下规定的包衣材料进行包衣和打光。
七、丸剂外观应圆整均匀、色泽一致。
蜜丸应细腻滋润,软硬适中。
蜡丸表面应光滑无裂纹,丸内不得有蜡点和颗粒。
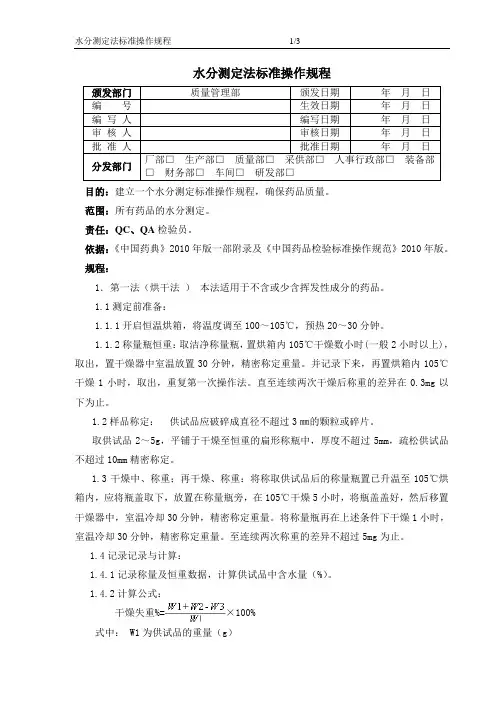
水分测定法标准操作规程颁发部门质量管理部颁发日期年月日编号生效日期年月日编写人编写日期年月日审核人审核日期年月日批准人批准日期年月日分发部门厂部□生产部□质量部□采供部□人事行政部□装备部□财务部□车间□研发部□目的:建立一个水分测定标准操作规程,确保药品质量。
范围:所有药品的水分测定。
责任:QC、QA检验员。
依据:《中国药典》2010年版一部附录及《中国药品检验标准操作规范》2010年版。
规程:1.第一法(烘干法)本法适用于不含或少含挥发性成分的药品。
1.1测定前准备:1.1.1开启恒温烘箱,将温度调至100~105℃,预热20~30分钟。
1.1.2称量瓶恒重:取洁净称量瓶,置烘箱内105℃干燥数小时(一般2小时以上),取出,置干燥器中室温放置30分钟,精密称定重量。
并记录下来,再置烘箱内105℃干燥1小时,取出,重复第一次操作法。
直至连续两次干燥后称重的差异在0.3mg以下为止。
1.2样品称定:供试品应破碎成直径不超过3㎜的颗粒或碎片。
取供试品2~5g,平铺于干燥至恒重的扁形称瓶中,厚度不超过5mm,疏松供试品不超过10mm精密称定。
1.3干燥中、称重;再干燥、称重:将称取供试品后的称量瓶置已升温至105℃烘箱内,应将瓶盖取下,放置在称量瓶旁,在105℃干燥5小时,将瓶盖盖好,然后移置干燥器中,室温冷却30分钟,精密称定重量。
将称量瓶再在上述条件下干燥1小时,室温冷却30分钟,精密称定重量。
至连续两次称重的差异不超过5mg为止。
1.4记录记录与计算:1.4.1记录称量及恒重数据,计算供试品中含水量(%)。
1.4.2计算公式:干燥失重%=×100%式中: W1为供试品的重量(g)W2为称瓶恒重的重量(g)W3为(称瓶+供试品)恒重的重量(g)。
1.5结果与判定:同烘干法。
1.6注意事项:用烘干法测定水分时,往往几个样品同时进行,因此称量瓶宜先用适宜的方法编码标记,瓶与瓶盖的编码一致,称量瓶放入烘箱的位置,取出冷却、称重的顺序,应先后一致。
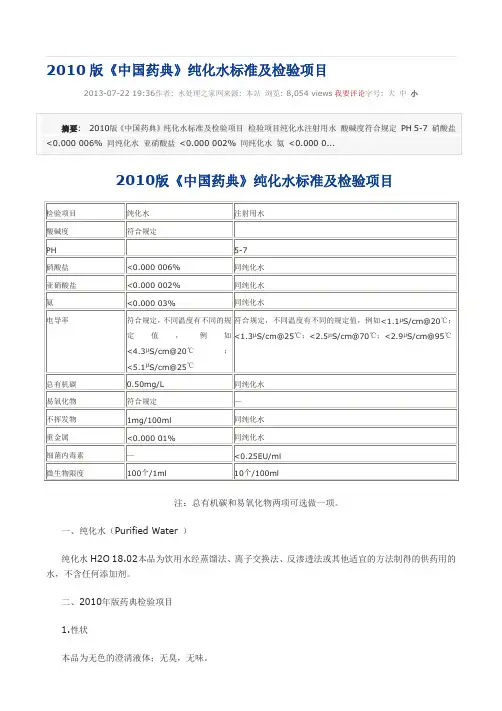
2010版《中国药典》纯化水标准及检验项目2013-07-22 19:36作者: 水处理之家网来源: 本站浏览: 8,054 views我要评论字号: 大中小注:总有机碳和易氧化物两项可选做一项。
一、纯化水(Purified Water )纯化水H2O 18.02本品为饮用水经蒸馏法、离子交换法、反渗透法或其他适宜的方法制得的供药用的水,不含任何添加剂。
二、2010年版药典检验项目1.性状本品为无色的澄清液体;无臭,无味。
取本品10ml,加甲基红指示液2滴,不得显红色;另取10ml,加溴麝香草酚蓝指示液5滴,不得显蓝色。
3.硝酸盐取本品5ml置试管中,于冰浴中冷却,加10%氯化钾溶液0.4ml与0.1%二苯胺硫酸溶液0.1ml,摇匀,缓缓滴加硫酸5ml,摇匀,将试管于50℃水浴中放置15分钟,溶液产生的蓝色与标准硝酸盐溶液[取硝酸钾0.163g,加水溶解并稀释至100ml,摇匀,精密量取1ml,加水稀释成100ml,再精密量取10ml,加水稀释成100ml,摇匀,即得(每1ml相当于1μgNO3)]0.3ml,加无硝酸盐的水4.7ml,用同一方法处理后的颜色比较,不得更深(0.000 006%)。
4.亚硝酸盐取本品10ml,置纳氏管中,加对氨基苯磺酰胺的稀盐酸溶液(1→100)1ml及盐酸萘乙二胺溶液(0.1→100)1ml,产生的粉红色,与标准亚硝酸盐溶液[取亚硝酸钠0.750g(按干燥品计算),加水溶解,稀释至100ml,摇匀,精密量取1ml,加水稀释成100ml,摇匀,再精密量取1ml,加水稀释成50ml,摇匀,即得(每1ml相当于1μgNO2))0.2ml,加无亚硝酸盐的水9.8ml,用同一方法处理后的颜色比较,不得更深(.0000 02%)。
5.氨取本品50ml,加碱性碘化汞钾试液2ml,放置15分钟;如显色,与氯化铵溶液(取氯化铵31.5mg,加无氨水适量使溶解并稀释成1000ml)1.5ml,加无氨水48ml与碱性碘化汞钾试液2ml制成的对照液比较,不得更深(0.000 03%)。

附件:《中国药典》2010年版(三部)凡例、通则及附录定稿会会议纪要按照2010年版《中国药典》编制的统一安排,我委于2009年3月18-20日在京召开了2010年版《中国药典》三部凡例、通则及附录的定稿会。
来自病毒制品、细菌制品、血液制品、生物技术产品以及微生物专业委员会的相关委员、中检所和参与批签发的7个地方药检所的有关专家、我委生物制品标准处、业务综合处相关人员以及部分生物制品生产企业代表共约40人参加了会议。
会议对2010年版《中国药典》三部凡例、9个通则及16个通用性附录的增修订进行了审定,确认了下列增修订意见,会议主要内容纪要如下:一、凡例(一)、名称及编排项下,增订微生态活菌制品总论及体外诊断试剂的收载。
(二)、设施与生产质量管理项下第(2),修订为:人血液制品应使用专用设备并在专用设施内进行生产,不得与其他异种蛋白制品混用。
(三)“制造”项修订为“基本要求”,修订内容为:1、设施与生产质量管理项下增订:(4)涉及感染性材料的操作应符合国家生物安全的相关规定。
2、辅料及原料项下将“原料”修订为“原材料”,质量要求增订应符合现行《中国药典》三部的规定,“本版药典未收载者,必须制定符合药用要求的标准”修订为“本药典未收载者,必须制定符合产品生产和质量控制要求的标准”。
3、增订“七、生产过程中防腐剂使用的相关要求”项,增订内容为:1、抗生素的使用生产过程中抗生素的使用应符合以下原则:(1)除另有规定外,不得使用青霉素或其他β-内酰胺类抗生素。
(2)成品中不得使用抗生素作为防腐剂。
(3)生产过程中,应尽可能避免使用抗生素,必须使用时,应选择安全性风险相对较低的抗生素,且产品的后续纯化工艺应保证可有效去除制品中的抗生素,去除工艺应经验证;如后续工艺不能有效去除抗生素,则不得添加。
病毒性疫苗生产中应仅限于在细胞制备、细胞增殖过程中使用抗生素。
(4)生产过程中使用抗生素时,成品检定中应检测抗生素残留量,并规定残留量限值。

中国药典2010版第一部
(原创版)
目录
1.引言:介绍《中国药典》及其 2010 年版的基本信息
2.《中国药典》2010 年版的主要内容
3.第一部的特点与重要性
4.第一部的主要章节和规定
5.结论:总结《中国药典》2010 年版第一部的主要贡献和影响
正文
《中国药典》是我国药品和药用辅料、直接接触药品的包装材料和容器的法定标准,是药品研发、生产、经营、使用和监管的重要依据。
2010 年版是该药典的第五版,其第一部是其中最重要的部分,涵盖了中药的基本要求、质量标准和检验方法等内容。
第一部的主要内容包括中药的来源、性状、鉴别、检查、浸出物测定、含量测定等。
其中,来源主要规定了中药材的采收、加工、贮存等环节,以保证其质量;性状则详细描述了中药的外观、气味、味道、水试、火试等特征,以便鉴别真伪;鉴别主要通过宏观和微观特征,如形状、大小、颜色、质地、组织结构等,以及理化和生物学特性,如旋光性、折光率、硬度、黏度、PH 值、酶谱、DNA 指纹等,来判断中药的真伪和品质;检查则规定了中药的重金属、农药残留、真菌毒素、杂质等污染物的限量,以保障用药安全;浸出物测定和含量测定则是评价中药有效成分含量的重要方法。
第一部的特点在于其全面性和严谨性。
全面性体现在它对中药的全过程进行了规定,从源头到成品,从质量到安全,涵盖了中药的各个方面;严谨性则体现在它对中药的鉴别和检查等环节的规定上,采用了现代科技手段,使得中药的质量控制更加科学和准确。
第一部的主要章节有中药的来源、性状、鉴别、检查、浸出物测定、含量测定等,规定了中药的基本要求、质量标准和检验方法。
其中,鉴别章节是第一部的重点,因为它是判断中药真伪和品质的关键。

2010版中国药典凡例总则一、《中华人民共和国药典》简称《中国药典》,依据《中华人民共和国药品管理法》组织制定和颁布实施。
《中国药典》一经颁布实施,其同品种的上版标准或其原国家标准即同时停止使用。
《中国药典》由一部、二部、三部及其增补本组成,内容分别包括凡例、正文和附录。
除特别注明版次外,《中国药典》均指现行版《中国药典》。
本部为《中国药典》二部。
二、国家药品标准由凡例与正文及其引用的附录共同构成。
本部药典收载的凡例、附录对药典以外的其他中药国家标准具同等效力。
三、凡例是为正确使用《中国药典》进行药品质量检定的基本原则,是对《中国药典》正文、附录及与质量检定有关的共性问题的统一规定。
四、凡例和附录中采用的“除另有规定外”这一用语,表示存在与凡例或附录有关规定不一致的情况时,则在正文中另作规定,并按此规定执行。
五、正文中引用的药品系指本版药典收载的品种,其质量应符合相应的规定。
六、正文所设各项规定是针对符合《药品生产质量管理规范》(Good Manufacturing Practices, GMP)的产品而言。
任何违反GMP 或有未经批准添加物质所生产的药品,即使符合《中国药典》或按照《中国药典》没有检出其添加物质或相关杂质,亦不能认为其符合规定。
七、《中国药典》的英文名称为Pharmacopoeia of The People’s Republic of China, 英文简称Chinese Pharmacopoeia;英文缩写为Ch.P.。
正文八、正文系根据药物自身的理化与生物学特性,按照批准的处方来源、生产工艺、贮藏运输条件等所制定的、用以检测药品质量是否达到用药要求并衡量其质量是否稳定均一的技术规定。
九、正文项下根据品种和剂型不同,按顺序可分别列有:(1)品名(包括中文名称、汉语拼音与英文名);(2)有机药物的结构式;(3)分子式与分子量;(4)来源或有机药物的化学名称;(5)含量或效价规定;(6)处方;(7)制法;(8)性状;(9)鉴别;(10)检查;(11)含量或效价测定;(12)类别;(13)规格;(14)贮藏;(15)制剂等。

比武试题九一、填空1.抗生素微生物检定包括两种方法,即管碟法和浊度法。
2.在抗生素微生物检定法中,二剂量法标准品溶液的高浓度所致的抑菌圈直径应在 18~22 ㎜。
3.在抗生素微生物检定法中,实测效价与估计效价:试验计算所得效价应在估计效价的±10% 以内。
4.抗生素微生物检定法常用检定菌为枯草芽孢杆菌、短小芽孢杆菌和藤黄微球菌。
5.抗生素微生物检定法系在适宜条件下,根据量反应平行线原理设计,通过检测抗生素微生物的抑制作用,计算抗生素活性(效价)的方法。
6.《中国药典》2010年版细菌内毒素检定法分凝胶法和光度测定法;后者包括浊度法和显色基质法。
7.凝胶法细菌内毒素检查用水系指内毒素含量小于 0.015 EU/ml的灭菌注射用水。
光度测定法用的细菌内毒素检查用水,其内毒素含量小于 0.005 EU/ml。
8.当细菌内毒检查测定结果有争议时,除另有规定外,以凝胶法为准。
9.无菌检查应在环境洁净度 10000 级下的局部洁净度 100 级的单向流空气区域或隔离系统中进行,其全过程必须严格遵守无菌操作,防止微生物污染。
10.药品进行无菌检查时所用的冲洗液有0.1%蛋白胨水溶液和pH7.0氯化钠-蛋白胨缓冲液。
11.无菌检定法有薄膜过滤法和直接接种法两种方法;细菌培养温度为32.5℃±2.5℃,真菌培养温度为25.5℃±2.5℃。
12.培养基灵敏度检查所用菌株为金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、生孢梭菌、白色念珠菌、黑曲霉。
13.洁净室的温度和相对湿度应与药品生产工艺要求相适应。
无特殊要求时,温度应控制在18~26℃,相对湿度控制在45%~65%。
14.眼部给药制剂微生物限度检查除细菌数、霉菌数和酵母菌数外还包括金黄色葡萄球菌、铜绿假单胞菌和大肠埃希菌。
15.微生物限度检查中,细菌培养温度为30~35℃;霉菌、酵母菌培养温度为23~28℃;控制菌培养温度为35~37℃。

附录Ⅴ 分光光度法分光光度法是通过测定被测物质在特定波长处或一定波长范围内的吸光度或发光强度,对该物质进行定性和定量分析的方法。
常用的波长范围为:(1) 200~400nm 的紫外光区;(2)400~760nm 的可见光区;(3) 760~2500nm 的近红外光区;(4)2.5~25μm (按波数计为4000~ 400cm -1)的中红外光区。
所用仪器为紫外分光光度计、可见分光光度计(或比色计)、红外分光光度计或原子吸收分光光度计。
为保证测量的精密度和准确度,所用仪器应按照国家计量检定规程或本附录规定,定期进行校正检定。
单色光辐射穿过被测物质溶液时,在一定的浓度范围内被该物质吸收的量与该物质的浓度和液层的厚度(光路长度)成正比,其关系如下式:A=lg T 1=Ecl式中 A 为吸光度;T 为透光率;E 为吸收系数,采用的表示方法是%11cm E ,其物理意义为当溶液浓度为1%(g/ml ),液层厚度为1cm 时的吸光度数值;c 为100ml 溶液中所含被测物质的重量(按干燥品或无水物计算),g ; l 为液层厚度,cm 。
物质对光的选择性吸收波长,以及相应的吸收系数是该物质的物理常数。
当已知某纯物质在一定条件下的吸收系数后,可用同样条件将该供试品配成溶液,测定其吸光度,即可由上式计算出供试品中该物质的含量。
在可见光区,除某些物质对光有吸收外,很多物质本身并没有吸收,但可在一定条件下加入显色试剂或经过处理使其显色后再测定,故又称比色分析。
附录Ⅴ A 紫外-可见分光光度法仪器的校正和检定1.波长 由于环境因素对机械部分的影响,仪器的波长经常会略有变动、因此除应定期对所用的仪器进行全面校正检定外,还应于测定前校正测定波长。
常用汞灯中的较强谱线237.83nm,253.65nm,275.28nm,296.73nm,313.l6nm,334.15nm,365.02nm,404.66nm,435.83nm,546. 07nm与576.96nm;或用仪器中氘灯的486.02nrn与656.10nm谱线进行校正;钬玻璃在波长279.4nm,287.5nm,333.7nm,360.9nm,418.5nm,460.0nm,484.5nm,536.2nm与637.5nm 处有尖锐吸收峰,也可作波长校正用,但因来源不同或随着时间的推移会有微小的变化,使用时应注意;近年来,常使用高氯酸钬溶液校正双光束仪器,以10%高氯酸溶液为溶剂,配制含氧化钬(Ho2O3) 4%的溶液,该溶液的吸收峰波长为241.13nm,278.10nm,287.18nm,333.44nm,345.47nm,361.31nm,416.28nm,451.30nm,485.29nm,536.64nm和640.52nm。

中国药典2010版2010版中国药典一部word版电子书(共1026页)2010版中国药典中国药典2010年版二部word版电子书中国药典沿革1953年版(第一版) 1949年10月1日中华人民共和国成立后,党和政府十分关怀人民的医药卫生保健工作,当年11月卫生部召集在京有关医药专家研讨编纂药典问题。
1950年1月卫生部从上海抽调药学专家孟目的教授负责组建中国药典编纂委员会和处理日常工作的干事会,筹划编制新中国药典。
1950年4月在上海召开药典工作座谈会,讨论药典的收载品种原则和建议收载的品种,并根据卫生部指示,提出新中国药典要结合国情,编出一部具有民族化、科学化,大众化的药典。
随后,卫生部聘请药典委员49人,分设名词、化学药、制剂、植物药、生物制品、动物药、药理、剂量8个小组,另聘请通讯委员35人,成立了第一届中国药典编纂委员会。
卫生部部长李德全任主任委员。
1951年4月24日至28日在北京召开第一届中国药典编纂委员会第一次全体会议,会议对药典的名称、收载品种、专用名词、度量衡问题以及格式排列等作出决定。
干事会根据全会讨论的意见,对药典草案进行修订,草案于1952年底报卫生部核转政务院文教委员会批准后,第一部《中国药典》1953年版由卫生部编印发行。
本版药典共收载品种531种,其中化学药215种植物药与油脂类65种,动物药13种,抗生素2种,生物制品25种,各类制剂211种。
1957年出版《中国药典》1953年版增补本。
1963年版(第二版) 1955年卫生部组建第二届药典委员会,聘请委员49人,通讯委员68人,此届委员会因故未能开展工作。
1957年卫生部组建第三届药典委员会,聘请委员80人,药学专家汤腾汉教授为这届委员会主任委员(不设通讯委员),同年7月28日至8月5日在北京召开第一次全体委员会议,卫生部李德全部长做了药典工作报告,特别指出第2010版中国药典pdf版(共三部,400多M)2010版中国药典一部word版电子书(共1026页)一版《中国药典》未收载广大民众习用的中药的缺陷。

执业药师中药学知识一备考试题:中药质量标准和判定(有答案)一、最正确选择题1、中药的安全性检查中,外源性有害物质不包含A、吡咯里西定B、黄曲霉毒素C、农药残留量D、重金属及有害元素E、二氧化硫残留量2、当前中药含量测定使用最多的方法是A、原子汲取分光光度法B、气相色谱法C、分光光度法D、原子发射光谱法E、高效液相色谱法3、显微鉴识时确认淀粉粒应加A、间苯三酚试液B、水合氯醛试液C、稀甘油D、盐酸E、碘试液4、“狮子盘头”所形容的药材是A、川木香B、党参C、北柴胡D、防风E、牛黄5、察看荧光时,若无特别说明,往常是指紫外光波长为A、254nmB、265nmC、365nmD、2540nmE、3650nm6、解离组织制片的氢氧化钾法合用于察看A、薄壁组织占大多数,木化组织少或分别存在的样品B、质地坚硬的样品C、木化组织许多的样品D、木化组织群集成束的样品E、叶类药材7、鉴识木质化细胞壁的试液是A、钌红试液B、氯化锌碘试液C、三氯化铁试液D、α- 萘酚试液E、间苯三酚试液和盐酸8、用显微鉴识法鉴识粘液采纳的试剂是A、碘试液B、苏丹Ⅲ试液C、钌红试液D、硝酸汞试液E、氯化锌碘试液9、拥有简单、易行、快速的特色的鉴识方法是A、基源判定B、性状判定C、显微判定D、理化判定E、含量测定10、制作解离组织制片刻,硝铬酸法合适于A、薄壁组织占大多数的样品B、木化组织少的样品C、木化组织分别的样品D、叶类、花类样品E、样质量地坚硬,木化组织许多或集成较大群束11、称取“ 0.1g ”系指称取重量可为A、0.06 ~0.14gB、0.01 ~0.1gC、0.05 ~0.15gD、0.1 ~0.15gE、0.05 ~0.1g12、《中国药典》 2010 年版规定,恒重是指供试品连续两次干燥或炽灼后称重的差别在() 的重量A、0.2mg 以下B、0.1mg 以下C、0.3mg 以下D、1.0mg 以下E、0.5mg 以下13、称取“ 2.00g ”系指称取重量可为A、1.995 ~2.005gB、2.005 ~2.995gC、1.095 ~2.005gD、1.995 ~2.995gE、2.00 ~2.005g二、配伍选择题1、A. 含挥发性成分的药品B.合用于各样成分的药材C.合用于果实种子类药材D.含挥发性成分的名贵药材E.不含或少含挥发性成分的药品<1> 、《中国药典》中水分测定的烘干法合用于ABCDE<2> 、《中国药典》中水分测定的甲苯法合用于ABCDE<3> 、《中国药典》中水分测定的减压干燥法合用于ABCDE2、A. 加硫酸无变化B.加稀盐酸溶解,同时有气泡发生C.间苯三酚试液再加盐酸显红色D.加苏丹Ⅲ试液搁置或微热显红色E.10%的α- 萘酚乙醇液再加浓硫酸显紫红色并很快溶解<1> 、角质化细胞壁ABCDE<2> 、硅质化细胞壁ABCDE<3> 、菊糖ABCDE<4> 、钟乳体ABCDE3、A.粘液B.淀粉粒C.碳酸钙结晶D.菊糖E.草酸钙结晶<1> 、哪项检查用钌红试液,显红色ABCDE<2> 、哪项检查用10%α- 萘酚乙醇溶液再加硫酸,显紫红色并很快溶解ABCDE4、A.狮子头B.蚯蚓头C.油头D.马头蛇尾瓦楞身E.云头<1> 、海马的外形如ABCDE<2> 、防风根头部习称ABCDE<3> 、党参根头部习称ABCDE5、A. 高效液相色谱法B.气相色谱法C.薄层色谱法D.红外光谱法E.二乙基硫代氨基甲酸银法<1> 、《中国药典》规定检测黄曲霉素的方法为ABCDE<2> 、《中国药典》规定检测农药残留的方法为ABCDE<3> 、《中国药典》规定检测砷盐的方法为ABCDE<4> 、《中国药典》规定检测二氧化硫残留量的方法为ABCDE三、综合剖析选择题1、主要存在于马兜铃属植物中的肾毒性成分马兜铃酸,最近几年来在国际上惹起激烈反响,引起人们对中药安全性的重视。
1.下列药品标准中属于法定标准的是()。
D 国家药典2.银量法测定巴比妥类药物,终点指示的原理是()。
B 过量1滴的滴定剂使巴比妥类形成难溶性的二银盐沉淀3.细菌内毒素检查采用的方法()。
B 鲎试剂法4.减少分析测定中偶然误差的方法为()。
B 增加平行试验次数5.原料药的含量测定宜选()。
C 容量分析法6.非水碱量法测定生物碱的硝酸盐()。
E 电位法指示终点7.中国药典规定紫外测定中,溶液的吸光度应控制在()。
C 0.3~0.78.具有怎样结构的甾体激素类药物可用四氮唑比色法测定()。
B C17-a- 醇酮基9.药品稳定性考察中加速试验的条件和所需样品批次是()。
C 40℃、RH75%,6个月,3批样10.下列哪一项不属于色谱系统适用性试验内容?() A 准确度11.拖尾因子表示()。
E 峰对称性12.荧光分析法适合于()。
D 低浓度溶液的测定13.一个药品的质量标准的主要内容包括()。
C 性状、鉴别、检查、含量测定14.澄清度检查()。
D 比浊度法15.维生素C中铜的检查()。
E 原子吸收分光光度法16.亚硝酸钠滴定法中,加KBr的作用是()。
E 生成NOBr以加速反应17.由信噪比S/N=10测得的最低浓度或量()。
B 定量限18.甲苯咪唑中无效晶型的检查()。
A 红外分光光度法19.在碱性水溶液中被铁氰化钾氧化后溶于正丁醇显蓝色荧光的药物是()。
B 维生素B120.对于毒性或贵重药材,应尽量采用()。
C 显微鉴别法21.盐酸去氧肾上腺素的溴量法原理()。
A 取代反应22.色谱法适合于多组分的分离分析,中药鉴别中最常用的方法是()。
E 薄层色谱法23.实验条件下被检杂质的适宜浓度,重金属为()。
E 0.01~0.02mg24.拆分酮康唑外消旋体的方法()。
B 手性流动相添加法25.用高氯酸滴定液直接滴定硫酸奎宁,1摩尔硫酸奎宁需消耗几摩尔高氯酸()。
附录IX H水分测定法测定用的供试品,一般先破碎成直径不超过3mm的颗粒或碎片;直径和长度在3mm 以下的可不破碎;减压干燥法需通过二号筛。
第一法(烘干法)本法适用于不含或少含挥发性成分的药品。
测定法取供试品2~5g,平铺于干燥至恒重的扁形称量瓶中,厚度不超过5mm,疏松供试品不超过10mm,精密称定,打开瓶盖在100~105℃干燥5小时,将瓶盖盖好,移置干燥器中,冷却30分钟,精密称定,再在上述温度干燥1小时,冷却,称重,至连续两次称重的差异不超过5mg为止。
根据减失的重量,计算供试品中含水量(%)。
第二法(甲苯法)本法适用于含挥发性成分的药品。
测定法取供试品适量(约相当于含水量1~4ml),精密称定,置短颈圆底烧瓶中,加甲苯约200ml,必要时加入干燥、洁净的沸石或玻璃珠数粒,将仪器各部分连接,自冷凝管顶端加入甲苯,至充满水分测定管的狭细部分。
将短颈圆底烧瓶置电热套中或用其他适宜方法缓缓加热,待甲苯开始沸腾时,调节温度,使每秒钟馏出2滴。
待水分完全馏出,即测定管刻度部分的水量不再增加时,将冷凝管内部先用甲苯冲洗,再用饱蘸甲苯的长刷或其他适宜的方法,将管壁上附着的甲苯推下,继续蒸馏5分钟,放冷至室温,拆卸装置,如有水黏附在水分测定管的管壁上,可用蘸甲苯的铜丝推下,放置,使水分与甲苯完全分离(可加亚甲蓝粉末少量,使水染成蓝色,以便分离观察)。
检读水量,并计算供试品中的含水量(%)。
【附注】用化学纯甲苯直接测定,必要时甲苯可先加水少量,充分振摇后放置,将水层分离弃去,经蒸馏后使用。
第三法(减压干燥法)本法适用于含有挥发性成分的贵重药品。
减压干燥器取直径12cm左右的培养皿,加入五氧化二磷干燥剂适量,使铺成0.5~1cm 的厚度,放入直径30cm的减压干燥器中。
测定法取供试品2~4g,混合均匀,分取约0.5~1g,置已在供试品同样条件下干燥并称重的称量瓶中,精密称定,打开瓶盖,放入上述减压干燥器中,减压至2.67kPa(20mmHg)以下持续半小时,室温放置24小时。
中国药典(2010年版)含量测定项中对照品配制、含量范围的研究摘要】目的在涉及到使用对照品的含量测定时,只有科学合理地配制对照品,使其配制的浓度准确可信,其药品的含量测定结果才准确可信。
方法对中国药典关于对照品的配制进行了认真分析,对比。
发现对照品配制有两种方法,一种是严格规定对照品称样量的具体数值,另一种是对照品称样量的数值相对不确定。
结果,通过分析大量的药品检验报告书实例,由于对照品的称样量数值不明确具体,发现检验员在实际操作中使用方法多样,具体是称样量多样,导致了稀释转移多样,影响了配制的对照品浓度的准确性。
结论明确对照品具体的称样量数值,明确具体稀释转移方法,所有对照品的配制都应规范到这一种方法中来。
【关键词】中国药典含量测定对照品配制《中国药典》由于在药品执法中的重要作用,其具有的科学性,权威性,可操作性,是我们药品检验工作的规范。
在对《中国药典》(2010年版一部:收载中药材及中成药)含量测定项的研究中,发现对照品的使用很多,这是一个好现象。
使用对照品(或标准品)进行对照检查,得出结论。
被认为是科学,客观,准确,可信度高。
是不让伪劣药品逃避技术监督进入市场的好方法。
用对照品检查含量,2010年版药典通常使用两种方法。
一种方法是从称样量到稀释转移直到最后结果,药典都明确规范,科学严谨,具有很强可操作性。
二部药典(收载化学及抗生素药品)大部分使用此方法。
我们从本版药典的一个实例来具体说明。
《中国药典》(2010年版一部)P47:马钱子,对照品溶液的制备:取士的宁对照品6mg,马钱子碱对照品5mg,精密称定,分别置10ml量瓶中加三氯甲烷使溶解并稀释至刻度,摇匀,分别精密量取2ml置同一10ml量瓶中,用甲醇稀释至刻度,摇匀,即得。
上述表达,重要的是称样量明确,稀释转移明确。
我们说药典的科学性,可操作性就表现在这里。
但是一部药典的药品中象这样配制对照品溶液的品种并不多。
多数是用另一种方式配制对照品溶液,其通常制备方式如下:称取XX对照品适量,精密称定,用XX试剂配制成含Xug/1ml的溶液,即得。
水分测定法
测定用的供试品,一般先破碎成直径不超过3mm的颗粒或碎片;直径和长度在3mm以下的可不破碎;减压干燥法需通过二号筛。
第一法(烘干法)本法适用于不含或少含挥发性成分的药品。
测定法取供试品2 ~ 5 g , 平铺于干燥至恒重的扁形称量瓶中,厚度不超过5mm,疏松供试品不超过l0mm,精密称定,打开瓶盖在100~105t干燥5小时,将瓶盖盖好,移置干燥器中,冷却30分钟,精密称定,再在上述温度干燥1小时,冷却,称重,至连续两次称重的差异不超过5mg为止。
根据减失的重量,计算供试品中含水量(%)。
第二法(甲苯法)本法适用于含挥发性成分的药品。
仪器装置如图。
A为5 0 0 m l的短颈圆底烧瓶;B为水分测定管;C为直形冷凝管,外管长40cm。
使用前,全部仪器应清洁,并置烘箱中烘干。
图甲苯法仪器装置
测定法取供试品适量(约相当于含水量l~4ml),精密称定,
置A瓶中,加甲苯约200ml,必要时加人干燥、洁净的沸石或玻璃珠数粒,将仪器各部分连接,自冷凝管顶端加人甲苯,至充满B管的狭细部分。
将A瓶置电热套中或用其他适宜方法缓缓加热,待甲苯开始沸腾时,调节温度,使每秒钟馏出2滴。
待水分完全馏出,即测定管刻度部分的水量不再增加时,将冷凝管内部先用甲苯冲洗,再用饱蘸甲苯的长刷或其他适宜的方法,将管壁上附着的甲苯推下,继续蒸馏5分钟,放冷至室温,拆卸装置,如有水黏附在B管的管壁上,可用蘸甲苯的铜丝推下,放置,使水分与甲苯完全分离(可加亚甲蓝粉末少量,使水染成蓝色,以便分离观察)。
检读水量,并计算供试品中的含水量(%)。
【附注】用化学纯甲苯直接测定,必要时甲苯可先加水少量,充分振摇后放置,将水层分离弃去,经蒸馏后使用。
第三法(减压干燥法)本法适用于含有挥发性成分的贵重药品。
减压干燥器取直径12cm左右的培养皿,加入五氧化二磷干燥剂适量,使铺成0. 5~lcm的厚度,放人直径30cm的减压干燥器中。
测定法取供试品2~4g,混合均匀,分取约0. 5~lg,置已在供试品同样条件下干燥并称重的称量瓶中,精密称定,打开瓶盖,放人上述减压干燥器中,减压至 2. 67kPa(20mmHg)以下持续半小时,室温放置24小时。
在减压干燥器出口连接无水氯化钙干燥管,打开活塞,待内外压一致,关闭活塞,打开干燥器,盖上瓶盖,取出称量瓶迅速精密称定重量,计算供试品中的含水量(%)。
五氧化二磷和无水氯化钙为干燥剂,干燥剂应
及时更换。
第四法(气相色谱法)
色谱条件与系统适用性试验用直径为0 . 18~0. 25mm的二乙烯苯-乙基乙烯苯型高分子多孔小球作为载体,柱温为 140~150’C,热导检测器检测。
注入无水乙醇,照气相色谱法(附录VI E)测定,应符合下列要求:
(1) 理论板数按水峰计算应大于1000,理论板数按乙醇峰计算应大于150;
(2) 水和乙醇两峰的分离度应大于2;
(3) 用无水乙醇进样5次,水峰面积的相对标准偏差不得大于3.0%。
对照溶液的制备取纯化水约0.2g,精密称定,置25ml量瓶中,加无水乙醇至刻度,摇匀,即得。
供试品溶液的制备取供试品适量(含水量约0.2g),剪碎或研细,精密称定,置具塞锥形瓶中,精密加人无水乙醇 50ml,密塞,混匀,超声处理20分钟,放置12小时,再超声处理 20分钟,密塞放置,待澄清后倾取上清液,即得。
测定法取无水乙醇、对照溶液及供试品溶液各1~5^1,注人气相色谱仪,测定,即得。
【附注】(1)对照溶液与供试品溶液的配制须用新开启的同一瓶无水乙醇。
(2)用外标法计算供试品中的含水量。
计算时应扣除无水乙醇中的含
水量,方法如下:
对照溶液中实际加人的水的峰面积=对照溶液中总水峰面积一KX对照溶液中乙醇峰面积
供试品中水的峰面积=供试品溶液中总水峰面积一K X供试品溶液中乙醇峰面积
K = 无水乙醇中水峰面积/无水乙醇中乙醇峰面积。