配位化学论文---分子轨道理论
- 格式:docx
- 大小:605.79 KB
- 文档页数:9

分子轨道理论的发展及其应用北京师范大学段天宇学号201111151097摘要:分子轨道理论是目前发展最成熟,应用最广泛的化学键理论之一。
本文简述了分子轨道理论的基本思想及发展历程,列举了其在配位化学、矿物学、气体吸附领域的应用实例,并对其前景作出展望。
0 前言化学键是化学学科领域中最为重要的概念之一。
通常,化学键被定义为存在于分子或晶体中或两个或多个原子间的,导致形成相对稳定的分子或晶体的强相互作用。
从二十世纪初期至今,科学家们为了解释化学键现象相继提出了价键理论、分子轨道理论、配位场理论等化学键理论。
其中分子轨道理论(Molecular Orbital Theory)具有容易计算、计算结果得到实验支持的优势,并不断得到完善与拓展,因而自二十世纪五十年代以来,已经逐渐确立了其主导地位[1]。
目前,作为相对最为成熟的化学键理论,分子轨道理论的应用已经涵盖了化学研究的几乎全部领域中。
1 分子轨道理论发展1926至1932年,Mulliken和Hund分别对分子中的电子状态进行分类,得出选择分子中电子量子数的规律,提出了分子轨道理论[2]-[3]。
分子轨道理论认为,电子是在整个分子中运动,而不是定域化的。
他们还提出了能级相关图和成键、反键轨道等重要概念。
1929年,Lennard-Jones提出原子轨道线性组合(Linear Combination of Atomic Orbitals)的理论[4]。
后来,原子轨道线性组合的思想被应用于分子轨道理论中,成为分子轨道理论的基本原理。
这一原理指出,原子轨道波函数通过线性组合,即各乘以某一系数相加得到分子轨道波函数。
这种组合要遵循三个基本原则,即:组合成分子轨道的原子轨道必须对称性匹配;组成分子轨道的原子轨道须能级相近;原子轨道达到最大程度重叠以降低组成分子轨道的能量。
其中,最重要的是对称性匹配原则,对称性相同的原子轨道组合成能量低于自身的成键分子轨道,对称性相反的原子轨道组合成高于自身的反键分子轨道。

摘要: (2)关键词: (2)Abstract: . (2)Keywords: (2)1.价键理论(Valence Bond Theory ) (2)2.晶体场理论CFT(Crystal Field Theory) (3)3.分子轨道理论((Molecular Orbital Theory) (4)4.配位场理论LFT (Ligand Field Theory) (4)5.经典配位化学的产生和发展 (5)6.配位化学新的发展及应用趋势 (6)7.配位化学近几年的研究热点 (6)8.结语 (6)参考文献 (6)浅谈配位化学理论摘要:自从1893年瑞士化学家维尔纳创立配位化学已来,配位化学理论得到不断发展,逐渐完善。
经过化学家们100多年的努力,由传统经典的配合物,发展到今天的配位超分子化合物,并显示出结构和功能上的优越特性,成为现代无机化学的一个发展方向。
本论文先对各配位理论进行简要的介绍,然后再总结其中的规律,最后根据发展的规律对未来的发展进行展望。
关键词:配位化学晶体场理论配位Abstract:since 1893,the Swiss chemist Werner has founded the coordination chemistry,coordination chemistry theory of continuous development,and gradually improve. After100years of efforts of chemists,from the traditional classic complexes,to today's development of supramolecular coordination compounds,and show the structural and functional superiority,becomes the modern inorganic chemistry is a direction of development. This paper first on the coordination theory was briefly introduced, and then summarizes the laws, according to the law of the development of the future development prospect.Keywords: Coordination chemistry Crystal field theory coordinatio n在阐明配位化合物结构的理论中,较重要的有价键理论、晶体场理论、分子轨道理论和配位场理论等,下面概述这些理论的基本内容:1.价键理论(Valence Bond Theory )价键理论是在Pauling 离子晶体电价规则基础上发展起来的, 它继承了电价规则中/原子的价分配在原子所连诸键上0的基本概念, 同时允许原子所连诸键的键价做不均匀的分配。

化学中的分子轨道理论化学是一门研究物质性质、组成及变化的科学,其中一个重要的方面是了解分子的构成和化学键的形成。
分子轨道理论是一个用于解释分子结构和化学键形成的重要理论。
在本文中,我们将深入探讨分子轨道理论的基本概念和应用。
分子轨道理论的基本概念分子轨道理论将分子看作是由原子轨道之间形成的新的轨道而构成。
原子轨道是一种描述电子位置的数学函数,它们描述了单个原子中电子的可能位置和能量。
但是,在两个或多个原子共同存在的分子中,原子轨道就发生了重叠,而由此形成了新的分子轨道。
有两种类型的分子轨道:成键分子轨道和反键分子轨道。
成键分子轨道是由原子轨道之间重叠形成的,这种重叠是化学键形成的原因。
反键分子轨道是由原子轨道不重叠的区域形成的,它们和成键分子轨道几乎具有相等的能量,但是它们的电子不会在化学键形成过程中参与,因此它们被称为反键分子轨道。
分子轨道理论的应用分子轨道理论可以用于解释分子的性质和化学反应。
让我们以氢分子为例,探讨分子轨道理论是如何解释氢分子的存在和相互作用的。
氢原子的电子结构是1s,其中一个s轨道中有一个电子。
当两个氢原子形成一个分子时,它们的s轨道相互重叠并形成了两个新的分子轨道:成键分子轨道和反键分子轨道。
成键分子轨道比原子轨道更稳定,因为它们的波函数符号相同,从而促进电子的互相吸引。
相反,反键分子轨道比成键分子轨道更不稳定,因为它们的波函数符号相反,在这种情况下,电子之间会互相排斥。
由于成键分子轨道比反键分子轨道更稳定,氢分子的所有电子都处于成键分子轨道中。
这样,它们就形成了共价键,并达到了更稳定的电子结构。
这就解释了为什么氢分子是存在的,而单个氢原子不会稳定存在。
分子轨道理论还可以用于预测化学反应的速率和化学键的强度。
它可以通过计算分子轨道重叠的程度来预测键的稳定性和长度。
此外,在有机化学中,分子轨道理论可以解释的许多现象,如亲电性、电子云和取代反应。
总结分子轨道理论是一个重要的化学理论。


分子轨道理论简介文章来源:/newsite/Web%20Page/GeneralChem/kechengneirong/09/9-4-1.htm价键理论着眼于成键原子间最外层轨道中未成对的电子在形成化学键时的贡献,能成功地解释了共价分子的空间构型,因而得到了广泛的应用。
但如能考虑成键原子的内层电子在成键时贡献,显然更符合成键的实际情况。
1932年,美国化学家Mulliken RS和德国化学家Hund F 提出了一种新的共价键理论——分子轨道理论(molecular orbital theory),即MO法。
该理论注意了分子的整体性,因此较好地说明了多原子分子的结构。
目前,该理论在现代共价键理论中占有很重要的地位分子轨道理论的要点1.原子在形成分子时,所有电子都有贡献,分子中的电子不再从属于某个原子,而是在整个分子空间范围内运动。
在分子中电子的空间运动状态可用相应的分子轨道波函数ψ(称为分子轨道)来描述。
分子轨道和原子轨道的主要区别在于:(1)在原子中,电子的运动只受1个原子核的作用,原子轨道是单核系统;而在分子中,电子则在所有原子核势场作用下运动,分子轨道是多核系统。
(2)原子轨道的名称用s、p、d…符号表示,而分子轨道的名称则相应地用σ、π、δ…符号表示。
2.分子轨道可以由分子中原子轨道波函数的线性组合(linear combination of atomic orbitals,LCAO)而得到。
几个原子轨道可组合成几个分子轨道,其中有一半分子轨道分别由正负符号相同的两个原子轨道叠加而成,两核间电子的概率密度增大,其能量较原来的原子轨道能量低,有利于成键,称为成键分子轨道(bonding molecular orbital),如σ、π轨道;另一半分子轨道分别由正负符号不同的两个原子轨道叠加而成,两核间电子的概率密度很小,其能量较原来的原子轨道能量高,不利于成键,称为反键分子轨道(antibonding molecular orbital),如σ*、π*轨道。

亚铜离子配合物的稳定性及应用近年来.由于金属配合物在日常生活和工业上都有广泛的应用,尤其过渡金属对探索和研究药物分子抗菌、抗肿瘤的作用机制具有重要意义。
在催化、光学材料以及电学材料等方面具有新型功能的金属配合物的研究也受到人们的广泛关注。
通过这一个学期的学习,我对配位化学的基础知识有了很大程度的了解。
在即将走完配位化学的课堂学习历程时,我就亚铜离子配合物的的稳定性及应用进行整理。
亚铜离子的化合价为+1,与铜离子相比较为稳定,但由于离子半径过大,不能存在于水溶液中,在酸性条件下自我岐化,生成Cu2+和Cu单质亚铜离子和铜离子可以相互转化:一般亚铜在固相或高温下稳定(亚铜离子在水相中会发生歧化),二价铜在水相中最稳定(因为二价水合铜的水和能特别大,因而亚铜容易歧化转变成稳定的二价铜)。
在溶液中稳定亚铜的另一途径是形成配合物。
如果非氧化性酸中的因此与亚铜离子有较强的配位能力,则可以提高铜的还原性(降低铜的电极电位),进而生成亚铜配离子。
亚铜离子在遇到强酸时会自我氧化生成铜离子和铜单质,现象为生成红色沉淀和蓝色溶液。
一价铜Cu(I)化合物通常不稳定,易被氧化成二价铜Cu(II)化合物。
从电子结构来看,单质铜为全满和半充满状态3d铒s9,失去一个电子而形成3d9489的全满和全空状态,均为较稳定的状态;而Cu(II)的电子结构为3d94so,3d9既非全满亦非半充满或全空状态,因此,Cu(D应该比较稳定。
实际在形成配合物时,由于Cu(II)的极化力比Cu(I)大,能与配体形成稳定的配位键,一般形成配位键的数目亦较多,使体系能量降低较多,因而通常更多地却是形成较为稳定的Cu(II)配合物。
相反,Cu(I)所带的电荷比Cu(II)的少,半径比Cu(II)的大,因而其成键能力弱于Cu(II),所以获得较为稳定的Cu(I)的配合物也因此成为人们研究的焦点。
铜的配合物常常具有一定的催化活性。
而亚铜化合物纳米材料的合成与应用研究正得到人们的青睐旧。
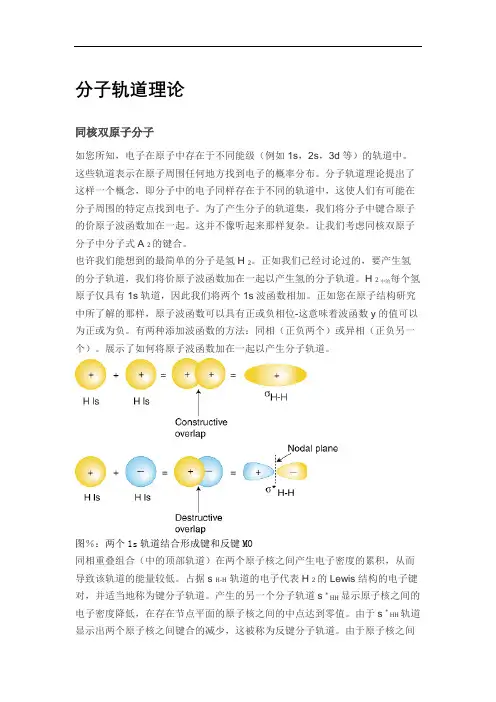
分子轨道理论同核双原子分子如您所知,电子在原子中存在于不同能级(例如1s,2s,3d等)的轨道中。
这些轨道表示在原子周围任何地方找到电子的概率分布。
分子轨道理论提出了这样一个概念,即分子中的电子同样存在于不同的轨道中,这使人们有可能在分子周围的特定点找到电子。
为了产生分子的轨道集,我们将分子中键合原子的价原子波函数加在一起。
这并不像听起来那样复杂。
让我们考虑同核双原子分子中分子式A 2的键合。
也许我们能想到的最简单的分子是氢H 2。
正如我们已经讨论过的,要产生氢每个氢的分子轨道,我们将价原子波函数加在一起以产生氢的分子轨道。
H 2中的原子仅具有1s轨道,因此我们将两个1s波函数相加。
正如您在原子结构研究中所了解的那样,原子波函数可以具有正或负相位-这意味着波函数y的值可以为正或为负。
有两种添加波函数的方法:同相(正负两个)或异相(正负另一个)。
展示了如何将原子波函数加在一起以产生分子轨道。
图%:两个1s轨道结合形成键和反键MO同相重叠组合(中的顶部轨道)在两个原子核之间产生电子密度的累积,从而导致该轨道的能量较低。
占据s H-H轨道的电子代表H 2的Lewis结构的电子键对,并适当地称为键分子轨道。
产生的另一个分子轨道s * HH显示原子核之间的电子密度降低,在存在节点平面的原子核之间的中点达到零值。
由于s * HH轨道显示出两个原子核之间键合的减少,这被称为反键分子轨道。
由于原子核之间电子密度的降低,抗键合轨道的能量高于键合轨道和氢1s轨道。
在分子H 2,没有电子占据反键轨道。
中总结这些关于键,反键和原子轨道的相对能量的发现,我们可以构建一个轨道相关图,如下所示:图%:氢的轨道相关图请注意,分离的原子的轨道写在图的两侧,是水平线,其高度表示它们的相对能量。
每个原子轨道上的电子用箭头表示。
在图的中间,写下了感兴趣分子的分子轨道。
虚线将母原子轨道与子分子轨道连接起来。
通常,键合分子轨道的能量低于其母原子轨道中的任何一个。


分子轨道理论和分子结构方法案例研究分子轨道理论和分子结构方法是现代化学领域中重要的研究内容,通过对分子的电子结构和化学键进行分析,能够揭示分子的性质和行为。
本文将介绍分子轨道理论和分子结构方法的基本原理,并通过详细的案例研究来加深我们对这两个领域的理解。
一、分子轨道理论分子轨道理论是研究分子中电子结构和性质的重要工具。
该理论是基于量子力学原理,通过数学方法描述分子中电子的运动和分布。
分子轨道理论假设分子中的每个电子都存在于特定的分子轨道中,这些分子轨道由分子中的原子轨道组合而成。
以分子氢(H2)为例,分子轨道理论可以解释H2的共价键形成。
两个氢原子靠近时,其原子轨道重叠,形成了分子中的分子轨道。
分子轨道可分为成键轨道(bonding orbitals)和反键轨道(antibonding orbitals)。
成键轨道由原子轨道同相叠加形成,能量较低,电子在其中较为稳定。
反键轨道由原子轨道异相叠加形成,能量较高,电子在其中较不稳定。
通过分子轨道理论,我们可以预测和解释许多分子的性质,如分子的磁性、反应活性和光谱行为等。
这使得分子轨道理论成为研究化学反应和分子性质的重要工具。
二、分子结构方法分子结构方法通过实验手段测定分子中原子的相对位置,从而揭示分子的结构和构型。
常用的分子结构方法包括X射线衍射、核磁共振(NMR)和质谱等。
1. X射线衍射X射线衍射是一种通过衍射现象确定晶体结构的方法,也被应用于分子结构的研究。
在X射线衍射实验中,X射线通过晶体或分子样品,与原子间的电子发生相互作用,形成衍射图样。
通过对衍射图样的解析,可以得到分子的结构信息,如原子之间的距离和角度等。
2. 核磁共振(NMR)核磁共振是一种研究分子结构和化学环境的强大工具。
通过对分子中原子核的核磁共振信号进行分析,可以确定分子的结构和化学键。
核磁共振技术广泛应用于有机化学、生物化学和医学领域,并已成为解析分子结构的重要方法之一。
3. 质谱质谱是一种通过测量分子或化合物中各个原子的质量进行分析的手段。

分子轨道理论简介我们把原子通过共用电子对结合的化学键成为共价键(covalent bond)。
路易斯(G.N.Lewis)曾经提出原子共用电子对成键的概念,也就是俗称的“八隅律”(高中阶段也只是停留于此)然而,我们知道很多现实情况都无法用八隅率解释,包括:PCl5,SCl6分子。
更重要的是,八隅率从来没有本质上说明共价键的成因:为什么带负电荷的两个分子不会排斥反而是互相配对?随着近代的量子力学(quantum mechanics)的建立,近代形成了两种现代共价键理论,即是:现代价键理(valence bond theory)简称VB(又叫作电子配对法)以及分子轨道理论(molecular orbital theory)简称MO。
价键理论强调了电子对键和成键电子的离域,有了明确的键的概念。
也成功的给出了一些键的性质以及分子结构的直观图像。
但是在解释H2+氢分子离子的单电子键的存在以及氧分子等有顺磁性或者大∏键的某些分子结构时感到困难。
而分子轨道理论可以完美的进行解释,这里我就主要阐述MO法的相关理论。
洪特(Hund)和密里肯(R.S Mulliken)等人提出了新的化学键理论,即是分子轨道理论。
这是人们利用量子力学处理氢分子离子而发展起来的。
(一)氢分子离子的成键理论氢分子离子(H2+)是由两个核以及一个电子组成的最简单分子,虽然不稳定,但是确实存在。
如何从理论上说明氢分子离子的形成呢?分子轨道理论把氢分子离子作为一个整体处理,认为电子是在两个氢核a和b组成的势场当中运动。
电子运动的轨道既不局限在氢核a的周围,也不会局限于氢核b的周围,而是遍及氢核a和b。
这种遍及分子所有核的周围的电子轨道,成为“分子轨道”。
如何形成这样的分子轨道呢?我们必须通过波函数来描述原子当中的运动状态,而波函数是薛定谔方程的解。
因为得到精确的薛定谔方程的解很困难,因此我们才取了近似方法,假设分子轨道是各个原子轨道的组成。
仍然以氢分子离子为例:当这个单电子出现了一个氢原子核a附近时候,分子轨道Ψ很近似于一个院子轨道Ψa。

配位化学论⽂⼩作业之酞菁配合物论⽂酞菁及其配合物的发展研究与应⽤摘要:本⽂介绍了酞菁化合物的发展简史;综述了酞菁的多种合成⽅法;对酞菁化合物在光电导体,⾮线性光学,发光,和有机超晶格结构等⽅⾯的应⽤和存在的问题作了详细描述;并对⾦属酞菁配合物的合成⽅法和应⽤(如,在癌症治疗⽅⾯)作了简要说明;同时也阐述了酞菁三明治的发展、合成和应⽤前景。
关键字:酞菁、合成、配合物、应⽤1.酞菁化合物的发展史l907年。
Braun等⼈在⼄醇中加热o-cyanobenzamide。
得到的⼀定数量的蓝⾊沉淀,后来证实这就是酞菁。
在三⼗年代早期,Linstead及其合作者合成了许多酞菁。
1935年,伦敦皇家学院的J. Monteath Robertson⽤升华法得到了可供X射线衍射研究的单晶,从⽽使酞菁成为第⼀个以X射线衍射⽅法被证实其分⼦结构特征的有机化合物。
酞菁环组成⼆维共轭π-电⼦体系,在此体系中,18个π-电⼦分别于内环C—N位[1],在红光区,酞菁具有强烈的吸收;其固态颜⾊依据中⼼原⼦,晶型,颗粒⼤⼩不同,可在深蓝⾊到⾦属铜和绿⾊之间变化。
由于酞菁是由van der waals构成的分⼦,存在各种各样的堆积⽅式,Iwatsu认为酞菁分⼦堆积是柱状平⾯结构,在⼀个酞菁柱内,其作⽤⼒主要来⾃第⼀临近位。
由于酞菁化合物的热稳定性(在空⽓中加热到400-500℃不发⽣明显分解),加上酞菁化合物种类的多样性和其表现出的优异性能,使得酞菁的基础和应⽤研究得以⼴泛进⾏。
在⼯业上,酞菁化合物已经⼴泛应⽤于染料和⾊素但是,酞菁化合物最近在其它领域也引起了⼴泛兴趣如能量转换(光伏打和太阳能电弛),光电导材料,⽓体检测,发光,光学⾮线性,光敏化荆(photosensitizers),整流器件(rectifying devices),光存储器件,液晶,低维材料和电致变⾊等。
这些应⽤⼤多与酞菁电⼦结构紧密有关。
对酞菁吸收谱研究表明酞菁有两个吸收带:⼀个是在600-700nm的可见光区(Q—band),另⼀个是在300-400nm 的近紫外光区(B-hand)。
晶体场理论与分子轨道理论的比较及配位场理论黄珺(湖北师范学院化学与环境工程系0303班,湖北黄石,435002)摘要:配位化合物中的化学键主要是指中心离子和配位体之间的化学键。
自1893年维尔纳提出了配位理论后,有关配合物中的化学键理论主要有现代价键理论、晶体场理论、配位键理论和分子轨道理论。
本文主要讨论分子轨道理论和晶体场理论。
分子轨道理论以量子力学为基础,用于说明共价分子结构。
晶体场理论是1929年由皮赛和范弗雷克提出的,用于配合物化学键研究,成功地解释了配合物的磁性、光学性质及结构等,故在配合物的化学键理论中确立了重要地位。
关键词:晶体场理论、分子轨道理论、配位场理论、配位键、化学键Crystal field theory and molecular orbit theory comparison and legend field theoryHuang Jun(Chemistry and environment engineering department, Hubei Normal University, Huangshi, 435002)Abstract:In the coordination compound chemical bond mainly was refers to between the central ion and the legend chemical bond .The Vyell natrium proposed since 1893 the coordinate theory ,in the related preparation chemical bond theory mainly had the present price key theory and the crystal field theory ,the coordination bond theory and the molecular orbit theory .This article main discussion molecular orbit theory and the crystal field theory .Molecular orbit theory take the quantum mechanics as a foundation ,used in explaining the covalent molecule structure .The crystal field theory was in 1929 proposes by H.Bathe and J.H.Van Vleck ,used in the preparation chemical bond research ,successfully explained and preparation magnetism ,the optical quality and the structure and so on ,therefore has established the important status in the preparation chemical bond theory. Key words: Crystal field theory molecular orbital theory legend field theory coordinate bond chemical bond晶体场理论是20世纪50年代初,在价键理论和纯静电理论的基础上发展起来的.晶体场理论把中心离子看作是带正电的点电荷,把配位体看作是带负电的点电荷,它们之间的结合完全看作是静电和排斥作用.同时考虑到配位体对中心离子d轨道的影响,它在解释光学和磁学等性质方面很成功.(分子轨道理论把组成分子的所有原子作为一个分子整体来考虑,在分子中的电子不再从属于某些特定的原子,而是遍及整个分子范围内运动,分子中每个电子运动状态,可以用波函数来描述.)[1]首先,来比较这两种理论的基本观点.晶体场理论的基本观点:(1) 在配合物中,中心离子和配位体之间的相互作用类似于离子晶体中正、负离子间的静电作用,故它们间的化学键力纯属静电作用力.(2)当中心离子(指d区元素的离子)处于由配体所形成的非球形对称的负电场中时,中心离子的d 电子将受到配体负电场的排斥作用,使5个等价的d轨道发生能级分裂,有些轨道的能量降低.(3)中心离子的d轨道产生能级分裂后,致使中心离子的d电子排布也发生变化,导致体系的能量变化.(分子轨道理论的基本观点是把分子作为一个整体加以考虑,而分子中的每个电子是这个整体中的一员,不再从属于原来所属的原子.第一,原子形成分子后,电子就不再局限于个别原子的原子轨道,而是从属于整个分子的分子轨道,所以分子轨道强调分子的整体性.第二,分子轨道中电子的分布也和原子中的电子分布一样,遵循保里不相容原理,能量最低原理和洪特规则,在分子轨道中电子可以配对,也可以不配对.第三,分子轨道中可以近似地通过原子轨道的适当组合而得到,分子轨道的数目等于组合前原子轨道数目之和,原子轨道在组合成分子轨道时,要符合分子对称原则,最大重叠原则和能量相近原则,才能形成有效的分子轨道.)[3]研究络合物结构就是研究络合物中配位体与中央金属之间的化学键.晶体场理论把M—L作用看作是不同对称性,正、负离子的静电作用,完全不考虑共价键的因素。
盛年不重来,一日难再晨。
及时宜自勉,岁月不待人。
盛年不重来,一日难再晨。
及时宜自勉,岁月不待人。
配位化学论文分子轨道理论的发展及其应用160113004 2013级化教一班马慧敏一、前言价建理论、分子轨道理论和配位场理论是三种重要的化学键理论。
三、四十年代,价键理论占主要的地位。
五十年代以来由于分子轨道理论容易计算且得到实验(光电能谱)的支持,取得了巨大的发展,逐渐占优势。
价建理论不但在理论化学上有重要的意义(下文中将详细介绍)。
在应用领域也有重要的发展,如分子轨道理论计算有机化合物的吸收光谱用于染料化学;前线分子轨道理论在选矿中的研究等等。
二、简介1、分子轨道理论产生和发展在分子轨道理论出现以前,价键理论着眼于成键原子间最外层轨道中未成对的电子在形成化学键时的贡献,能成功地解释了共价分子的空间构型,因而得到了广泛的应用。
但如能考虑成键原子的内层电子在成键时贡献,显然更符合成键的实际情况。
1932年,美国化学家 Mulliken RS和德国化学家HundF 提出了一种新的共价键理论——分子轨道理论(molecular orbital theory),即MO法。
该理论注意了分子的整体性,因此较好地说明了多原子分子的结构。
目前,该理论在现代共价键理论中占有很重要的地位。
以下是各个年代提出的关于分子轨道理论的一些重要理论和方法,是分子轨道理论发展过程中的几个里程碑!1926-1932年,在讨论分子光谱时,Mulliken和Hund提出了分子轨道理论。
认为:电子是在整个分子轨道中运动,不是定域化的。
他们还提出能级图、成键、反键轨道等重要的概念。
1931-1933年,Hukel提出了一种简单的分子轨道理论,用于讨论共轭分子的性质,相当成功。
1950年,Boys用Guass函数研究原子轨道,解决了多中心积分问题,是今天广为利用的自洽场分子轨道理论的基础,在量子化学的研究中占有重要地位。
1951年,Roothaan在Hartree-Fock方程的基础上,把分子轨道写成原子轨道的线性组合,得到Roothaan方程。
分子轨道理论及基态与激发态分子轨道理论基本概念一、分子轨道:(molecular orbital) 描述分子中电子运动的波函数,指具有特定能量的某电子在相互键合的两个或多个原子核附近空间出现的概率最大的区域。
分子轨道由原子轨道线性组合而成。
二、成键三原则:能量相近、最大重叠、对称性匹配。
只有对称性相同的两个原子轨道才能组成分子轨道。
σ对称:一个原子轨道,取X轴作为对称轴,旋转180°,轨道符号不变。
如S,Px,d x2-y2为σ对称。
π对称:一个原子轨道,取X轴作为对称轴,旋转180°,轨道符号改变。
Py,Pz,d xy是π对称。
由σ对称的原子轨道组成的键——σ键由π对称的原子轨道组成的键——π键三、成键轨道与反键轨道分子轨道与原子轨道的联系:轨道守恒——2个原子轨道线性组合,产生2个分子轨道;能量守恒——2个分子轨道的总能量等于2个原子轨道的总能量;能量变化——每个分子轨道的能量不同于原子轨道的能量组合结果—定会出现能量高低不同的两个分子轨道。
——这是原子轨道线性组合的方式不同所致。
波函数同号的原子轨道相重叠,原子核间的电子云密度增大,形成的分子轨道的能量比各原子轨道能量都低,成为成键分子轨道。
波函数异号的原子轨道相重叠,原子核间的电子云密度减小,形成的分子轨道的能量比各原子轨道能量都高,成为反键分子轨道。
四、电子填入分子轨道时服从以下原则:1、能量最低原理:电子在原子或分子中将优先占据能量最低的轨道。
2、保利不相容原理:在同一原子或分子中、同一轨道上只能有两个电子,且自旋方向必须相反。
3、洪特规则:在能量相同的轨道中(简并轨道),电子将以自旋平行的方式、分占尽可能多的轨道基态与激发态当分子中的所有电子都遵从构造原理的这三个原则时,分子所处的最低能量状态——基态。
通常情况下,分子处于基态。
激发态:当分子获取能量后,分子中的电子排布不完全遵从构造原理,分子处于能量较高的状态——激发态,是原子或分子吸收一定的能量后,电子被激发到较高能级但尚未电离的状态。
应用分子轨道理论处理双原子分子结构1101班阮赛摘要:分子轨道理论(MO理论)是处理双原子分子及多原子分子结构的一种有效的近似方法,是化学键理论的重要内容。
它注重于分子轨道的认知,即认为分子中的电子围绕整个分子运动,注意了分子的整体性,因此较好地说明了多原子分子的结构。
目前,该理论在现代共价键理论中占有很重要的地位。
本文就分子轨道理论介绍及应用于双原子分子的一些内容作简单介绍。
关键词:分子轨道理论、、成建轨道、反键轨道、对称性匹配、最大重叠、能量相近, σ键、π键、键级、顺磁性、反磁性等。
背景:从20世纪30年代初,由Hund,Mulliken,Lennard-Jones开创,Slater,Hückel,Pople发展至今。
该方法的分子轨道具有较普通的数学形式,较易程序化。
六十年代以来,随着计算机的发展,该方法得到了很大的发展。
如Pople等研制的Gaussian从头算程序, 已成为当今研究化学键理论的主流方法.一、分子轨道理论(Molecular orbital theory)要点1、分子轨道理论的基本观点是把分子看作一个整体,其中电子不再从属于某一个原子而是在整个分子的势力场范围内运动。
正如在原子中每个电子的运动状态可用波函数(ψ)来描述那样,分子中每个电子的运动状态也可用相应的波函数来描述。
2、分子轨道是由分子中原子的原子轨道线性组合而成,简称LCAO (linear combination of atomic orbitals 的缩写)。
组合形成的分子轨道数目与组合前的原子轨道数目相等。
如两个原子轨道ψa和ψb线性组合后形成两个分子轨道ψ1和ψ2ψ1 = c1ψa +c2ψb ;ψ2= c1ψa -c2ψb这种组合和杂化轨道不同,杂化轨道是同一原子内部能量相近的不同类型的轨道重新组合,而分子轨道却是由不同原子提供的原子轨道的线性组合。
原子轨道用s、p、d、f……表示,分子轨道则用σ、π、δ……表示。
配位化学论文分子轨道理论的发展及其应用160113004 2013级化教一班马慧敏一、前言价建理论、分子轨道理论和配位场理论是三种重要的化学键理论。
三、四十年代,价键理论占主要的地位。
五十年代以来由于分子轨道理论容易计算且得到实验(光电能谱)的支持,取得了巨大的发展,逐渐占优势。
价建理论不但在理论化学上有重要的意义(下文中将详细介绍)。
在应用领域也有重要的发展,如分子轨道理论计算有机化合物的吸收光谱用于染料化学;前线分子轨道理论在选矿中的研究等等。
二、简介1、分子轨道理论产生和发展在分子轨道理论出现以前,价键理论着眼于成键原子间最外层轨道中未成对的电子在形成化学键时的贡献,能成功地解释了共价分子的空间构型,因而得到了广泛的应用。
但如能考虑成键原子的内层电子在成键时贡献,显然更符合成键的实际情况。
1932年,美国化学家 Mulliken RS和德国化学家Hund F 提出了一种新的共价键理论——分子轨道理论(molecular orbital theory),即MO法。
该理论注意了分子的整体性,因此较好地说明了多原子分子的结构。
目前,该理论在现代共价键理论中占有很重要的地位。
以下是各个年代提出的关于分子轨道理论的一些重要理论和方法,是分子轨道理论发展过程中的几个里程碑!1926-1932年,在讨论分子光谱时,Mulliken和Hund提出了分子轨道理论。
认为:电子是在整个分子轨道中运动,不是定域化的。
他们还提出能级图、成键、反键轨道等重要的概念。
1931-1933年,Hukel提出了一种简单的分子轨道理论,用于讨论共轭分子的性质,相当成功。
1950年,Boys用Guass函数研究原子轨道,解决了多中心积分问题,是今天广为利用的自洽场分子轨道理论的基础,在量子化学的研究中占有重要地位。
1951年,Roothaan在Hartree-Fock方程的基础上,把分子轨道写成原子轨道的线性组合,得到Roothaan方程。
1952年,福井谦一提出前线分子轨道理论,用以讨论分子的化学活性和分子间相互作用等,可以解释许多实验结果。
1965年,Woodward和Hoffman提出分子轨道对称守恒原理,发展成讨论基元反应发生可能性的重要规则。
用于指导某些复杂化合物分子的合成。
2、分子轨道理论的含义和一些重要分子轨道的构成方法1)分子轨道理论的含义分子轨道理论(Molecular orbital theory),简称MO理论,是处理双原子分子及多原子分子结构的一种有效的近似方法,是化学键理论的重要内容。
它与价键理论不同,后者着重于用原子轨道的重组杂化成键来理解化学,而前者则注重于分子轨道的了解,认为:原子在形成分子时,所有电子都有贡献,分子中的电子不再从属于某个原子,而是在整个分子空间范围内运动。
[1]其基本观点是:物理上存在单个电子的自身行为,只受分子中的原子核和其他电子平均场的作用,以及泡利不相容原理的制约;数学上则企图将难解的多电子运动方程简化为单电子方程处理。
因此,分子轨道理论是一种以单电子近似为基础的化学键理论。
描写单电子行为的波函数称轨道(或轨函),所对应的单电子能量称能级。
对于任何分子,如果求得了它的系列分子轨道和能级,就可以像讨论原子结构那样讨论分子结构,并联系到分子性质的系统解释。
计算化学中常以原子轨道线性组合近似来计算分子轨道波函数:[2]式中的c ij系数可由将等式代入薛定谔方程以及应用变分原理求得。
简单地讲,该方法意即,分子轨道由原子轨道组合而成。
原子轨道波函数各乘以某一系数相加或相减,得到分子轨道波函数。
组合时原子轨道对分子轨道的贡献体现在系数上,组合前后轨道总数不变。
简单讲,分子轨道就是由一系列遵从能量相近原则、最大重叠原则、对称性匹配原则的原子轨道,通过一定方向重叠而形成的具有不同能量的一系列轨道。
相同相位重叠则能量比形成它的原子轨道能量低,为成键轨道,电子优先占据。
而以相反相位重叠而形成的轨道能量升高,在这些轨道上电子出现的几率小。
形成分子轨道后,所有的电子离域,按照能量最低原则,Hund规则,泡利不相容原则,在分子轨道中排布。
2)常用的构成分子轨道的方法不同形态的原子轨道按照一定的方向重叠,形成具有不同对称性的分子轨道。
各种分子轨道具有不同的对称性,可依此将其分为σ、π与δ三种类型。
上图虚线表示截面,可以看出三种不同轨道对称性的差别:•σ分子轨道:对键轴呈圆柱形对称,成键σ轨道如σg1s 为中心对称,反键σ轨道如σu1s 为中心反对称。
•π分子轨道:对平面xy 反对称,只有一个含键轴的节面,对节面呈反对称性。
•δ分子轨道:通过键轴节面的分子轨道,对两个节面都呈反对称性。
接下来,介绍原子轨道组成分子轨道的最常见的几种类型:最常见的σ分子轨道:可以由能量相近的两个s 轨道,s-p 轨道,p-p 轨道头碰头重叠而成。
如下图所示:常见的∏轨道:两个p 轨道肩并肩重叠形成两个π分子轨道。
其中能量较高的为πp *反键轨道较低的为πp 成键轨道两个s 轨道重叠产生两个σ分子轨道。
能量较高的为σ*反键轨道 能量低的为σ成键轨道两个p 轨道头碰头重叠产生两个σ分子轨道。
能量较高的为σ*反键轨道 能量低的为σ成键轨道s 轨道和Pz 轨道沿z 轴重叠产生两个σ分子轨道。
能量较高的为σ*反键轨道 能量低的为σ成键轨道.d xy与pz轨道对称性匹配,肩并肩重叠形成两个π轨道(如图为成键轨道,反键轨道未画出)两个d xy轨道对称性匹配,肩并肩重叠形成两个π轨道(如图为成键轨道,反键轨道未画出)δ分子轨道:两个d xy轨道沿z轴重叠成δ分子轨道三、分子轨道理论的应用及成就简单分子轨道理论的应用应用简单分子轨道理论,画出分子轨道能级图,确定电子排布,能够利用其进行很多定性和定量的研究。
如计算键级,判断多原子分子是否能稳定结合,研究双原子的成键方式,判断磁性等。
也可以用于计算键长,键解离能,双原子分子的偶极矩等。
例如:1.对于氧气成键状态和顺磁性的解释:对于第二周期同核双原子分子,不考虑s-p杂化时分子轨道能级图如下图所示,能级顺序为2.分子轨道理论在生物无机化学中也有广泛的应用。
生物无机化学主要研究一些金属离子与生物配体(蛋白质、氨基酸、核酸等)配位,形成生物活性物质,而发挥催化、调节等作用。
而分子轨道理论常用于解释一些配位的机理,从而发挥作用。
对于研究配合物的性质,作用机理,以及指导人工合成某些模拟化合物有重要的意义。
N2是生物固氮的原料,与CO是等电子体。
它们具有相似的价电子组态,和成键状态。
然而CO与金属活性中心的配位作用远比N2强。
用分子轨道理论能够解释这一现象。
N 2和CO的分子轨道能级顺序不同于O2,由于它们原子轨道重叠形成的σs, σp轨道的能量相近,对称性匹配,能进一步组合成新的分子轨道,因此能级位置发生变化。
(对第二周期元素:Li, Be, O, F用简单分子轨道能级顺序;N, C, B 需要考虑s-p杂化。
)以下是N2、CO的分子轨道能级图根据能量最低,以及Pauli不相容原则。
O2的12个价电子组态为(σ2s)2(σ2s*)2(σ2pz)2(π2px)2(π2py)2(π2px*)1(π2py*)1,键级=2, 其中6个p电子,形成两个三电子π键。
在π2px*和π2py*轨道分别有一个未成对电子,自旋相同。
很好地说明O2分子的顺磁性。
N2的轨道能级图N 2和CO 都是10个价电子, 组态为(1σg )2(2σu )2(1πu )4 (3σg )2.成键情况相似。
在与金属配位时,3σg 上一对电子进入金属的空轨道,形成σ配位键;同时过渡金属的d 电子进入2π反键轨道,形成反馈π键,从而构成σ-π协同配位结构。
同CO 相比,N 2最高占据轨道能量比较低,所以N 2是一个比较差的电子给予体,给出电子形成σ配位键的能力较弱;另一方面,N 2最低未占据空轨道能量比CO 高,所以N 2接受d 电子形成反馈π键的能力也不如CO 强。
因此,N 2分子配合物的稳定性比金属羰基化合物差。
将其应用于生物化学中,探寻能更好的与N 2配位的催化剂,而实现人工固氮,具有远大的应用价值。
Hukel 分子轨道理论的应用(HMO 法) Hukel 分子轨道理论的基本思想是:a) 把电子间的双粒子相互作用近似得用单电子平均位场代替,从而导致分子体系单电子运动方程b)c) 引入Hukel 近似d) 由于π轨道和σ轨道的对称性不同,不能混合,可以将其分别处理。
对于共轭分子将原子核、内层电子、σ电子看作分子骨架,只考虑外部的π电子。
Hukel 分子轨道理论能够很好得解释共轭分子的稳定性。
通过Hukel 分子轨道理论能用两个参数α、β表示分子轨道所对应各个能级的能量。
能够很好得解释共轭分子的稳定性。
通过每个电子的波函数算出分子中每个原子的电荷密度、相邻原子π键级、自由价、定域能、前线轨道指数等参数能够预测共轭分子的化学反应活性,和反应类型。
例如:① 如下图所示利用Hukel 分子轨道理论做出标有电荷密度、相邻原子π键级、自由价的分子图,可以判断甘菊环2、3位置容易发生亲电取代,4,6位置易发生亲核取代。
CO 的轨道能级图适用于第二周期异核双原子分子②HMO理论对于解释环状共轭分子的稳定性有重要的贡献。
证明了4N+2规则:含有4n+2个π电子的单环共轭体系是稳定的,而含有4n个π电子的共轭单环不稳定。
对于含有4n+2个π电子的环状分子,所有的电子都会处于成键轨道;而对于含有4n个π电子的共轭单环,有两个电子会占据二重简并的两个非键轨道,容易发生结构畸变,简并消除,所以是不稳定的。
证明过程在这里就不详细说明了。
含有不同电子数目的单环共轭体系的分子轨道能级图③HMO法还可以用于合成一些有趣的共轭大分子。
例如1984年,SYoneda等人合成了一个由由两个三元环两个五元环构成的,容易形成如下图b构型而稳定的有机分子。
用HMO法计算表明:与这个分子相似的n1环与n2环像下图中一样在一个大环上交替出现组成共轭体系,则在n1+n2=4n时,分子稳定。
前线分子轨道理论的应用前线分子轨道理论强调分子的性质,特别是分子与其它分子的相互作用主要是由最高占据轨道HOMO轨道,和最低未占据轨道LOMO轨道所决定的。
前线分子理论指出分子在反应过程中必须遵从一定的原则一个分子的HOME和另一个分子的LOMO轨道必须对称性一致,并且相同相位能够重叠。
互相作用的HOME和LOMO轨道能量接近。
随两个分子HOME和LOMO轨道发生叠加,电子从一个分子的HOME转移到另一个分子的LOMO轨道,电子转移方向从电负性判断要合理。
利用前线分子轨道理论可以判断化学反应能否发生以及发生的难易程度。
以及反应条件。
前线轨道给出了分子互相作用时,电子流动的简单图像(在满足以上规则的情况下,HOME和LOMO轨道之间能量差小的,是主要电子流动方向)。