【淋巴瘤专家共识 2018】原发中枢神经系统淋巴瘤的诊断与治疗
- 格式:doc
- 大小:15.41 KB
- 文档页数:6



中枢神经系统淋巴瘤,中枢神经系统淋巴瘤症状中枢神经系统淋巴瘤包括原发中枢神经系统的淋巴瘤和全身淋巴瘤侵入中枢神经系统的继发性淋巴瘤。
以前命名很多,现称其为淋巴瘤或恶性淋巴瘤。
中枢神经系统淋巴瘤的病因(一)发病原因中枢神经系统内无淋巴循环及淋巴结,对淋巴瘤的病因目前有三种学说。
早期认为淋巴瘤起源于软脑膜血管的膜周细胞,后侵入邻近脑组织,并扩展到穿支血管周围间隙,侵犯半球深部结构。
第二种学说认为,淋巴瘤是非肿瘤性淋巴细胞在中枢神经系统反应性集聚所致。
由于脑组织缺乏淋巴系统,单核炎症细胞数相对为低,脑组织的免疫功能相对较薄弱,在慢性抗原的刺激下,免疫系统以多克隆形式反应,当抗原进一步刺激而淋巴细胞增生时,可能发生特异性的基因突变,形成单克隆增殖而发展成恶性淋巴瘤。
第三种学说认为,淋巴结或淋巴结以外B淋巴细胞间变成肿瘤,肿瘤细胞随血循环迁移,因其细胞表面携带中枢神经系统特异吸附标记物,故仅聚集于中枢神经系统,而真正的原发部位却不清楚,此学说可以解释颅内多发淋巴瘤。
(二)发病机制淋巴瘤可发生在中枢神经系统的任何部位,但大多数发生在幕上,大约50%的淋巴瘤发生在大脑半球,后颅窝占10%~30%,幕上、下同时受累占18%,病变好发于基底神经节、胼胝体、脑室周围白质和小脑蚓部,软脑膜、脉络丛和透明隔也常受累。
脑内淋巴瘤可为局灶性占位病变或弥散性浸润生长,肿瘤绝无包膜。
局灶性占位可多发,常位于脑室旁,为实体性病变,边界不清,周围水肿明显,质地可软、可硬,血运丰富,灰白色或紫红色,很少出血、坏死囊变。
弥散性生长的肿瘤大体观可正常,可有蛛网膜下腔扩张,致使其增厚呈灰白色,其属于B细胞型淋巴瘤,以小细胞型和大细胞型者多见。
镜下显示弥散性的肿瘤细胞浸润,远超出大体边界,细胞致密,胞质少,多呈圆形或卵圆形,细胞核明显,变长或扭曲,染色质多而分散,核分裂现象多见,有时瘤细胞呈套袖状沿血管周围分布,有时也可见肿瘤周围脑组织内呈巢灶状分布的肿瘤细胞,甚至远离肿瘤的脑组织内也可见到散在或簇状分布的肿瘤细胞,这可能构成肿瘤多中心性或复发的基础,肿瘤血运丰富,多属中等以下之小血管。

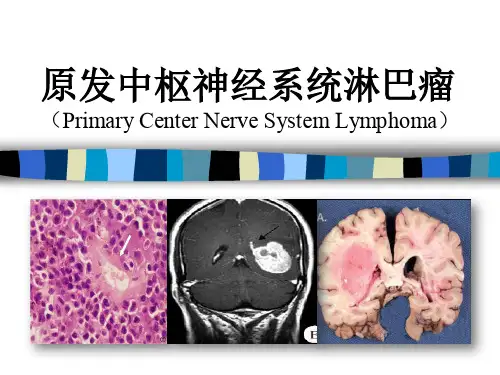
【淋巴瘤专家共识2018】原发中枢神经系统淋巴瘤的诊断与治疗原发中枢神经系统淋巴瘤原发中枢神经系统淋巴瘤(Primary central nervous system lymphoma,PCNSL)是原发于结外的非霍奇金淋巴瘤,仅限于颅脑、软脑膜、脊髓和眼球组织侵犯,而无其他组织或淋巴结浸润。
PCNSL是一种罕见的中枢神经系统肿瘤,约占颅内肿瘤的3%,占非霍奇金淋巴瘤中的1%,病理类型90%以上为弥漫大B细胞淋巴瘤,50%·70%BCL-6高表达,90%以上MUM-1高表达,具有高度侵袭性,预后差。
该病中位发病年龄60岁左右,且近年来在免疫功能正常的患者中发病率呈逐年上升趋势。
由于PCNSL的发病部位特殊及血脑屏障的存在,对于该病的诊断及治疗均存在困难,目前暂无标准治疗方案。
由于眼球组织侵犯,以局部治疗为主,不在此讨论。
PCNSL的影像学诊断PCNSL由于发病位置特殊,活检困难,影像学早期诊断十分重要。
目前标准的影像学诊断及疗效评估主要依赖T1加权增强磁共振成像(MRI)。
尽管免疫正常PCNSL患者中约50%在T1加权MRI上可见增强病灶,但有约25%的患者表现为T2/FLAIR上的高密度非增强病灶,因此T2/FLAIR有助于检测出一些无症状非增强的中枢神经淋巴瘤。
值得注意的是,由于放化疗及退行性变导致的脑白质病变亦可导致T2/FLAIR的脑白质变性,因此亟待新的影像学手段进一步提高诊断水平。
弥散加权是目前研究较多的一种影像学检测手段,有助于区分细胞较多弥散受限的PCNSL和少细胞的肿瘤如胶质瘤,也与预后相关。
Wieduwilt等发现低弥散系数患者预后更好。
代谢影像学如正电子发射计算机断层显像(PET)/MRI、波谱分析亦是新的研究方向。
PCNSL诊断标志物早诊断早治疗对于提高PCNSL患者预后至关重要,但由于PCNSL侵犯部位多为深部脑实质,活检较为困难。
研究人员对于PCNSL协诊标志物进行了研究,发现一些驱化因子(如CXCL13)和细胞因子(如IL-10)可以与影像学结合协助诊断。



原发性中枢神经系统淋巴瘤治疗及症状:原发性中枢神经系统淋巴瘤(primary central nervous system lymphoma,PCNSL)是一种仅发生于脑和脊髓,而没有全身其他淋巴结或淋巴组织浸润的淋巴瘤。
原发性中枢神经系统淋巴瘤的临床表现:原发性中枢神经系统淋巴瘤的临床表现很不一致,这主要与肿瘤的生长部位和范围有关。
大部分患者都有脑内病变的症状和体征。
由于PCNSL的病变50%位于额叶且为累及多叶,患者多表现为性格的改变,头痛,乏力,嗜睡。
癫痫发作比胶质瘤,脑膜瘤及脑内转移瘤的发生率低,这可能在明确诊断PCNSL时,病变较少累及易发生癫痫的脑皮质区有关。
此外,有的病人表现为幻觉、幻视、幻听等精神症状,亦可以表现为健忘,智力减退。
症状的持续时间为几周至几个月,这与PCNSL的预后差有关。
免疫缺陷患者的临床表现与免疫功能正常患者有所不同。
例如,AIDS患者多有精神智力方面的改变,多系统的缺损的改变,多并发其他疾病,如病毒性脑炎,弓形虫病,进行性多叶脑白质病等。
原发性中枢神经系统淋巴瘤治疗:对于原发性中枢神经系统淋巴瘤患者的治疗,主要有放射治疗,化学治疗,手术治疗。
据调查,对于PCNSL患者只进行手术治疗,预后和生存率并没有改善,故目前仅限于定向性诊断,和顽固性脑水肿的姑息治疗。
应用广泛的治疗手段为皮质激素、放疗和化疗。
化疗对部分免疫功能正常的PCNSL患者有一定的疗效,但是,如何明确何种病人对化疗有效,化疗方案的选择,鞘内注射的时机和剂量,放疗的剂量,药物的神经毒副作用等都有待于我们进一步的研究。
如病人的一般情况良好,医生建议可在原发性中枢神经系统淋巴瘤化疗的同时与cls细胞疗法相联合应用,还应加强全身支持治疗,纠正吸收不良综合征。

原发性中枢神经系统淋巴瘤的诊断与治疗原发性中枢神经系统淋巴瘤(primary central nervous system lymphoma,PCNSL)是指原发于脑、脊髓、眼或软脑膜的淋巴瘤,大多数 PCNSL 为 B 细胞起源,形态及病理特征与弥漫大B 细胞淋巴瘤(diffuse large B- cell lymphoma,DLBCL)相似,WHO (2008)造血与淋巴组织肿瘤分类已经将原发于中枢神经系统(CNS)的DLBCL 归类为一个独立实体。
PCNSL 占脑肿瘤的 3%,95% 以上为 DLBCL,好发于 50-70 岁者,起病至就诊时间多在 2-3 个月以内。
由于抗 HIV 药物的应用,继发于 HIV 感染者发病率有下降趋势,而无HIV 感染者的发病率有升高趋势。
PCNSL 患者主要表现为精神状态的改变、颅内压增高如头痛、恶心呕吐及视乳头水肿以及局部压迫症状,包括癫痫、记忆力减退、行走不稳、视野障碍、言语模糊以及轻度偏瘫。
除了脑部受累,还有10%-20% 患者有眼部受累,表现为视物模糊或者诉有“漂浮物”。
由于 T 细胞起源的以及其他类型的 PCNSL 发生率极低,且大多仅限于个案报道,故本文我们主要针对 CNS 的 DLBCL 的诊断和治疗进展进行综述。
一、PCNSL 的诊断(一)影像学颅脑影像学检查对于 PCNSL 临床诊断与鉴别诊断具有重要作用。
下面就几种主要检查方法进行介绍。
1.MRI:PCNSL 的MRI 特征是在TIWI 呈等或稍低信号,T2WI 呈稍低、等或高信号,单个或多个同质病变,较局限,边缘不规则,90% 病变周围伴有不同程度的水肿,通常能够接触到脑脊液表面,增强后肿瘤明显均匀一致增强是本病的特点,为血脑屏障破坏使对比剂渗透到细胞外间隙的结果。
坏死、边缘强化、出血及钙化并不常见。
在免疫缺陷患者,可见多发病灶呈环状强化。
60%-70% 的患者肿瘤为单发病灶,80%-90% 的病灶位于小脑幕上。

原发中枢神经系统淋巴瘤一、概述原发性中枢神经系统淋巴瘤(primary central nervous system lymphoma, PCNSL)是指患者诊断是肿瘤局限于大脑、小脑、脑干、眼、软脑膜和脊髓等中枢神经部位,尚未发现累及中枢神经系统以外的淋巴瘤。
PCNSL属于结外侵袭性非霍奇金淋巴瘤的一种特殊类型,占所有结外淋巴瘤4-6%,占所有原发CNS肿瘤的3%。
在PCNSL中,96%以上为高度恶性的B细胞淋巴瘤,弥漫大B细胞淋巴瘤是其中最常见的亚型,T细胞淋巴瘤和低度恶性淋巴瘤仅占1-4%。
二、病史体检1)年龄;有无重要脏器功能不全(主要指心、肝、肾功能)。
ECOG评分2)肿瘤的局部症状:重点询问有无神经、精神症状体征如认知缺损、偏瘫、语言障碍、记忆丧失、小脑症状及头痛、恶心、呕吐等非特异性颅内压增高的表现3) B症状:发热(重点询问发热的程度,热型,伴随症状),消瘦(近期体重下降程度),盗汗。
4)体格检查:重点皮肤检查;神经系统检查;眼部检查三、特异的实验室检测项目1)免疫组化:大部分病例是生发中心B细胞来源的,具有很高的免疫球蛋白基因突变发生率,VH4-34基因常常优先表达。
而且中枢神经系统大B细胞淋巴瘤还表达激活的外周B细胞型淋巴瘤的免疫分子表型标志物MUM1。
2)细胞遗传学:应用比较基因组杂交技术研究发现,在PCNSL常见的重现性基因组改变包括:1q,7q,12q和18q的增加,6q的缺失。
四、诊断由于PCNSL的诊断必须以组织学证据为基础,因此活检是必需的。
立体定向穿刺术既可实现活检病理组织,又可避免较大的手术创伤,是目前首选的有创诊断方法。
组织学诊断明确后,应进行更详细的检查以明确分期。
12.5%表现为局限于CNS病变的患者被发现有CNS外的损害,为了明确分期,建议全身CT扫描(胸、腹、盆腔)、骨髓穿刺及活检,老年男性患者需行双侧睾丸超声检查(因为睾丸淋巴瘤很容易侵犯CNS),眼部受累应进行眼前房、玻璃体、眼底检查。

原发性中枢神经系统淋巴瘤怎样治疗?*导读:本文向您详细介绍原发性中枢神经系统淋巴瘤的治疗方法,治疗原发性中枢神经系统淋巴瘤常用的西医疗法和中医疗法。
原发性中枢神经系统淋巴瘤应该吃什么药。
*原发性中枢神经系统淋巴瘤怎么治疗?*一、西医*1、治疗对于原发性中枢神经系统淋巴瘤患者的治疗,主要有放射治疗,化学治疗,手术治疗。
据调查,对于PCNSL患者只进行手术治疗,预后和生存率并没有改善,故目前仅限于定向性诊断,和顽固性脑水肿的姑息治疗。
应用广泛的治疗手段为皮质激素、放疗和化疗。
目前,术后放疗是原发性中枢神经系统淋巴瘤患者的常规治疗之一,平均生存时间为8~18个月。
曾有实验报道,单独手术治疗的生存时间与手术后放疗相比为4.6个月∶15.2个月。
同时,根据多个实验证实,PCNSL患者的生存率与放疗剂量有一定的关系。
1967年,Sagerman实验表明,放疗剂量低于30Gy的治疗全部失败,而部分病人的治疗剂量较大,却得到缓解。
Murray调查198例病人,54例剂量大于50Gy,其余病人小于50Gy,5年生存率前者为42.3%,后者为12.8%。
RTOG在1983~1987年调查表明,有41例免疫功能正常的患者进行了脑部放疗,其中脑和脑膜的放疗剂量为40Gy,瘤体的剂量为60Gy,并在瘤体周围增加了2cm的放疗范围。
治疗结果为,平均生存时间为11.6个月,48%的病人生存期为1年,28%为2年。
对原发性中枢神经系统淋巴瘤的患者同时进行脑和脊髓的放疗,目前有一定的争议。
虽然,软脑膜性PCNSL的发病率在上升,但是,目前并没有确切的实验证明此种放疗优于脑部放疗。
而且,同时进行脑和全身脊髓的大规模放疗,也损伤了脊椎和骨盆的大量骨髓储备,这对随后的全身化疗有一定的影响。
所以,除非有确切的脊髓内PCNSL证据,一般不采取脊髓的放疗。
虽然大剂量放疗对PCNSL患者有一定的疗效,但仍有部分患者对放疗无效或复发。
在前文所提到的RTOG调查中,41例中有3例在中枢系统外有复发,1例眼部病变未能缓解。
原发性中枢神经系统淋巴瘤的诊断和治疗:临床指南《Expert Rev Neurother》Schafer N原发性中枢神经系统淋巴瘤(PCNSLs)是淋巴结外的非何杰金氏淋巴瘤,96%以上的PCNSLs属于弥漫性大B细胞淋巴瘤,肿瘤的增殖指数大于50%,恶性程度很高。
在美国PCNSLs 发病率为4.6/100万,发病年龄以60-65岁为主。
罹患免疫缺陷疾病或接受免疫抑制治疗的人群,如:AIDS,器官移植者患病风险增高。
德国波恩大学医学院神经肿瘤科Schafer N教授等发表原发性中枢神经系统淋巴瘤的综述性文章,并提出临床指南意见,诊断:PCNSLs有认知损害、偏瘫、小脑症状和颅高压表现等等主要临床症状。
10%的患者因肿瘤累及眼眶出现视力障碍,其中90%可在数月至数年后发展至颅内,10%软脑膜受累。
该肿瘤在MRI的T1WI上为等或低信号,增强扫描为多发的均匀强化病灶,其边缘呈云雾状,常位于脑室旁或脑表面。
最终确诊依赖于组织病理学检查,免疫正常患者发现颅内多发病变怀疑PCNSLs时需要通过立体定向穿刺活检或脑脊液中分离得到肿瘤细胞进行组织病理学诊断。
由于激素可能会影响组织学诊断,因此活检前不可使用。
病理诊断后进一步做肿瘤分级。
此外,通过裂隙灯检查是否累及眼部;全身影像学检查、骨髓活检或FDG-PET判断颅外受累情况。
病程和预后相关因素仅仅以类固醇激素或支持治疗,PCNSLs患者的中位生存时间只有3个月;手术并不能延长生存期。
全脑放疗后生存期可以达到12-18月;积极的放化疗或化疗可使中位生存时间达到60月以上。
60岁以下的患者5年生存率接近70%。
大样本多变量分析发现,年龄>60岁和KPS评分是重要的预后相关因素。
而多发病灶、累积软脑膜与预后的相关性不大。
AIDS病人患PCNSLs,如无特殊治疗生存期少于2个月,半数以上死于机会性感染。
治疗原则:与系统性淋巴瘤不同,PCNSLs患者不适合采取标准的R-CHOP方案(环磷酰胺、阿霉素、长春新碱、泼尼松龙),可能与肿瘤细胞完整的血脑屏障保护有关。
中枢神经系统淋巴瘤是原发于中枢神经系统的淋巴瘤(PCNSL),本病少见,仅占中枢神经系统肿瘤的1%~3%。
约8%淋巴瘤原发于中枢神经系统,半数颅内淋巴瘤病人伴有全身淋巴瘤。
PCNSL可在任何年龄发病,男性略多于女性。
其来源不明,可发生在中枢神经系统内的各个部位,但大多数发生在幕上,尤以基底神经节、胼胝体、脑室周围白质和小脑蚓部好发,也可累及软脑膜、脉络丛和透明膈。
脑内淋巴瘤可为局灶性占位病变或弥散性侵润生长,无包膜。
局灶性占位可多发,边界不清,周围水肿明显。
质地可软可硬,血运丰富,但很少出血、坏死。
弥漫性生长的淋巴瘤一般为B细胞型淋巴瘤,以小细胞型和大细胞型多见,其大体观可正常。
淋巴瘤镜下示:弥散性肿瘤细胞侵润,远超过大体边界,细胞致密,胞浆少,多呈圆形或卵圆形,细胞核明显,染色质多而分散,核分裂现象多见,有时瘤细胞呈袖套样沿血管周围分布,有时也可见肿瘤周围组织内呈巢状分布的肿瘤细胞。
血运丰富,多属中等以下小血管。
PCNSL病程短,大多数在半年以内,主要症状与体征为其占位性病变或弥漫性脑水肿引起,比如头痛呕吐等颅高压症状,可伴有精神方面的改变。
局灶体征取决于肿瘤的部位和范围。
辅助检查,周围血像淋巴细胞可增高,但无特异性。
脑脊液中,几乎所有病人脑脊液中蛋白含量高,半数病人的脑脊液中能检出肿瘤细胞和淋巴细胞计数增高。
80%的病人的脑电图不正常,显示局灶性或弥散性病变。
头颅影像学无特异性表现。
由于该病临床表现和辅助检查不典型,所以常导致诊断困难,需与胶质母细胞瘤、转移瘤、脑膜瘤、脑脓肿和脑炎鉴别。
而最终的诊断有赖于立体定向活检。
本病的治疗应以综合治疗为主,以活检-化疗-放疗为最佳方案。
【淋巴瘤专家共识2018】原发中枢神经系统淋巴瘤的诊断
与治疗
原发中枢神经系统淋巴瘤原发中枢神经系统淋巴瘤(Primary central nervous system lymphoma,PCNSL)是原发于结外的非霍奇金淋巴瘤,仅限于颅脑、软脑膜、脊髓和眼球组织侵犯,而无其他组织或淋巴结浸润。
PCNSL 是一种罕见的中枢神经系统肿瘤,约占颅内肿瘤的3%,占非霍奇金淋巴瘤中的1%,病理类型90%以上为弥漫大B细胞淋巴瘤,50%·70%BCL-6高表达,90%以上MUM-1高表达,具有高度侵袭性,预后差。
该病中位发病年龄60岁左右,且近年来在免疫功能正常的患者中发病率呈逐年上升趋势。
由于PCNSL的发病部位特殊及血脑屏障的存在,对于该病的诊断及治疗均存在困难,目前暂无标准治疗方案。
由于眼球组织侵犯,以局部治疗为主,不在此讨论。
PCNSL的影像学诊断PCNSL由于发病位置特殊,活检困难,影像学早期诊断十分重要。
目前标准的影像学诊断及疗效评估主要依赖T1加权增强磁共振成像(MRI)。
尽管免疫正常PCNSL患者中约50%在T1加权MRI上可见增强病灶,但有约25%的患者表现为T2/FLAIR上的高密度非增强病灶,因此T2/FLAIR有助于检测出一些无症状非增强的中枢神经淋巴瘤。
值得注意的是,由于放化疗及退行性变导
致的脑白质病变亦可导致T2/FLAIR的脑白质变性,因此亟待新的影像学手段进一步提高诊断水平。
弥散加权是目前研究较多的一种影像学检测手段,有助于区分细胞较多弥散受限的PCNSL和少细胞的肿瘤如胶质瘤,也与预后相关。
Wieduwilt等发现低弥散系数患者预后更好。
代谢影像学如正电子发射计算机断层显像(PET)/MRI、波谱分析亦是新的研究方向。
PCNSL诊断标志物早诊断早治疗对于提高PCNSL患者预后至关重要,但由于PCNSL侵犯部位多为深部脑实质,活检较为困难。
研究人员对于PCNSL协诊标志物进行了研究,发现一些驱化因子(如CXCL13)和细胞因子(如IL-10)可以与影像学结合协助诊断。
同时也可在脑脊液中检测microRNA(21、19b、92a)等协助诊断。
但目前这些研究仍处于临床前阶段,需要大规模临床试验进一步证实。
PCNSL的治疗目前对于PCNSL,尚无标准治疗方案,国际上较为推崇的治疗方案分为两个阶段:诱导治疗及巩固治疗。
诱导治疗以大剂量甲氨蝶呤(HD-MTX,甲氨蝶呤>1 g/m2)为基石的治疗方案为主,但HD-MTX的具体剂量、输注时间亦暂无标准,较为常用的是MTX3.5 g/m2 3小时输注。
同时HD-MTX需与其他药物联合治疗,IELSG20临床试验已证实HD-MTX联合大剂量阿糖胞苷较之HD-MTX 单药CR及3年PFS均可显著提高(18%对46%,21%对
38%,p<0.05)。
目前对于HD-MTX需予何种药物联合运用,暂无定论。
IELSG32随机对比了HD-MTX+大剂量阿糖胞苷、HD-MTX+大剂量阿糖胞苷+利妥昔单抗、HD-MTX +大剂量阿糖胞苷+噻替派+利妥昔单抗三组疗效,证实在HD-MTX+大剂量阿糖胞苷基础上同时加用利妥昔单抗、噻替派显著提高客观缓解率(ORR)、2年总生存(OS)(40%对51%,42%对56%对59%)。
值得注意的是,在该研究中HD-MTX 大剂量阿糖胞苷基础上单纯加用利妥昔单抗,仅能提高ORR,而完全缓解(CR)、OS无明显差异。
但亦有报道利妥昔单抗可改善PCNSL中预后,需要进一步随机对照研究明确。
国际上目前对于年轻、体力状态好的患者,较为支持在HD-MTX基础上联合使用烷化剂、利妥昔单抗±大剂量阿糖胞苷。
对于无法耐受HD-MTX+大剂量阿糖胞苷的患者,亦可使用HD-MTX联合替莫唑胺、甲基苄肼等药物作为诱导治疗方案。
ANOCEF及GOELAMS 协作组报道HD-MTX 替莫唑胺作为诱导方案,ORR可达71%,1年无进展生存(PFS)率为36%,2年OS率为58%,副作用小,毒性可耐受。
巩固治疗主要以全颅放疗(WBRT)及自体造血干细胞移植为主,全颅放疗剂量≤50 GY,可有瘤区加强放疗。
G-PNSL-SG1临床试验对比了诱导治疗后CR患者观察或行WBRT,发现WBRT可提高PFS,但对OS无影响。
IELSG32发现在≤70岁的患者中无
论使用WBRT或清髓行自体造血干细胞移植,2年PFS无显著差异(80%对69%,P<0.05),2年OS率>80%。
值得注意的是,由于目前临床试验随访时间较短,对于WBRT带来的迟发性的神经系统损伤可能估计过低。
降低WBRT剂量可能可以减低WBRT所带来的神经系统损伤,但可能导致疾病进展。
因此,对于使用WBRT作为巩固治疗,尤其在老年人中,必须小心谨慎。
对于不适于使用WBRT及自体造血干细胞移植的患者,也可使用口服烷化剂(替莫唑胺、甲基苄肼等)维持治疗,Nordic研究小组使用替莫唑胺作为>65岁患者维持治疗(150 mg/m2,d1~5,每28天,维持1年),2年OS率为60%,提高疗效。
目前一些新的靶向药物在PCNSL治疗中也取得了不错的疗效。
2项临床试验检测了依鲁替尼在PCNSL中的疗效,ORR分别为55%及68%。
1项研究来那度胺在复发PCNSL中的治疗结果显示,ORR可达67%。
4名患者使用PD-1治疗复发PCNSL,ORR100%,目前疗效持续均已达1年以上。
推荐治疗前的检查治疗前的检查包括病史询问、体格检查(包括全身浅表淋巴结、韦氏环、肝脾、神经系统查体)、体能状态、B症状、血常规、生化常规、乙肝两对半(乙肝感染者查乙肝病毒载量)、HIV病毒检测、妊娠检测(育龄期妇女)、骨髓穿刺检查、骨髓活检检查、胸腹盆增强CT 或PET/CT、脊髓增强MRI(如脊髓有侵犯,脑脊液阳性)、
头颅增强MRI,眼MRI(如眼及其附属物有侵犯)、裂隙灯检查、脑脊液检查(如安全情况下)、睾丸B超(>60岁男性)。
由专业的肿瘤病理专家诊断或会诊组织病理,免疫组化指标包括:CD3、CD5、CD10、CD20、CD45、CD30、BCL2/6、IRF4/MUM1、MYC、Ki-67、ALK、CD19、CD38、CD21、CD138、CyclinD1、EBBER。
推荐的分期PCNSL局限于颅内,暂无具体分期。
推荐的预后评价系统国际结外淋巴瘤研究组(IESLG)标准:年龄>60 岁,美国东部肿瘤协作组(ECOG )评分>1 分,乳酸脱氢酶(LDH)升高,脑脊液蛋白升高,肿瘤位于脑深部(低危0~1、中危2~3、高危4~5)。
推荐的治疗方案诱导治疗方案推荐大剂量MTX为主的治疗方案:lMA±R方案:Rituxmab 375 mg/ m2 d0 ,HD-
MTX3.5 g/m2 3小时输注d1,阿糖胞苷2 g/m2 2次/天、1小时输注、d2~3 ,每14天一个疗程,共8个疗程lMT±R 方案:Rituxmab 375 mg/ m2 d0 ,HD-MTX 3.5 g/m2 3小时输注d1,替莫唑胺150 mg/m2,d1-5疗程2、4、6、8;每14天一个疗程,共8个疗程MPV±R方案:Rituxmab 375 mg/m2 d0 ,HD-MTX 3.5 g/m2 3小时输注d1,长春新碱1.4 mg/m2 d1,甲基苄肼100 mg/m2 d1~7,每14
天一个疗程,共8个疗程(如无法耐受大剂量甲氨蝶呤治疗方案,可考虑放疗或替莫唑胺单药治疗)
巩固治疗方案自体造血干细胞移植EA方案:VP-16 40 mg/kg 96小时持续输注;阿糖胞苷2 g/m2 2次/天d1-4替莫唑胺维持治疗:替莫唑胺150mg/m2,d1-5 每28天一个疗程共维持1年
推荐的治疗流程图PCNSL。