阿莫西林测定包合率
- 格式:doc
- 大小:78.00 KB
- 文档页数:3

实验十二阿莫西林胶囊的分析一、实验目的1.熟悉高效液相色谱仪的结构及正确使用。
2.掌握阿莫西林胶囊分析的原理及方法。
二、实验原理阿莫西林为β–内酰胺类抗生素,其母核部分与侧链部分均有共轭体系,有紫外吸收,因此阿莫西林胶囊的鉴别、有关物质的检查、含量测定均采用反相高效液相色谱法。
有关物质的检查方法为外标法和不加校正因子的主成分自身对照法,含量测定采用外标法定量。
三、实验方法本品含C16H19 N3O5S应为标示量的90.0%~110.0%。
【鉴别】在含量测定项下记录的色谱图中,供试品溶液主峰的保留时问应与对照品溶液主峰的保留时间一致。
【检查】水分取本品的内容物,照水分测定法(附录M第一法A)测定,含水分不得过16.0%。
有关物质取本品的内容物适量,精密称定,用流动相A溶解并制成每1ml中含2mg 的溶液,滤过,取续滤液照阿莫西林有关物质项下的方法测定。
单个杂质的峰面积不得大于对照溶液主峰面积的2倍(2.0%),各杂质峰面积的和不得大于对照溶液主峰面积的5倍(5.0%)。
供试品溶液中汪何小于对照溶液主峰面积0.05倍的峰可忽略不计)溶出度取本品,照溶出度测定法(附录X C第一法),以水900ml为溶出介质,转速为每分钟100转,依法操作,经45分钟时,取溶液适量,滤过,精密量取续滤液适量,用溶出介质稀释成缈"ml中约含130μg的溶液,照紫外一可见分光光度法(附录ⅣA),在272nm 的波长处测定吸光度;另取装量差异项下的内容物,混合均匀,精密称取适量(约相当于平均装量),按标示量加溶出介质溶解并稀释成每1ml中约含130μg的溶液,滤过,取续滤液作为对照溶液,同法测定,计算每粒的溶出量。
限度为80%,应符合规定。
其他应符合胶囊片剂项下有关的各项规定(附录I A)。
【含量测定】1.色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;以0.05mol/L磷酸二氢钾溶液(2mol/L氢氧化钠溶液调节PH值至5.0)–乙(97.5︰2.5)为流动相;流速为1ml/min,检测波长为254nm,理论板数按阿莫西林峰计算不低于2000。

阿莫西林制剂的含量测定技术【摘要】药物阿莫西林的原有剂型有片剂、胶囊剂等,目前又出现了一些新的剂型、制剂,为了适应新剂型、制剂的要求和进一步完善阿莫西林原有的质控标准,文章对测定阿莫西林制剂含量的方法进行了研究,并在本文中作了阐述。
【关键词】阿莫西林;测定技术作为光谱抗生素药物的一种,临床上阿莫西林被广泛应用,其又名为羟氨苄青霉素,属β-内酰胺青霉素类抗生素。
作为阿莫西林研究的一个重要方面,阿莫西林制剂的含量的测定方法一直备受关注。
文章对测定阿莫西林制剂含量的方法进行了研究。
测定阿莫西林制剂含量的测定方法主要有微生物效价测定法、碘量法、电位滴定法、旋光法、分光光度法以及流动注射化学发光法等,下面对以上这几种方法一一说明。
1.微生物效价测定法作为抗生素测定经典方法之一,微生物效价测定法同样适用于阿莫西林制剂含量的测定,通过该种方法测定出的数据能够直接反应阿莫西林对病菌微生物的抑杀能力,但该种方法的不足之处是实验操作繁琐、工作量较大。
具体方法是以藤黄八叠球菌(28001)为试验菌种,试验培养基为药典Ⅰ号培养基进行实验,最低检测浓度为0.05mg/L,日内和日间变异系数小于15%,线性范围为0.05~400mg/L(r=0.9977,P<0.01),符合生物样品分析要求。
2.碘量法碘量法是利用阿莫西林分子不消耗碘,而其降解产物消耗碘的特性来测定阿莫西林含量的方法,也是一种测定青霉素类抗生素较为通用的方法。
具体方法是先将阿莫西林制剂水解成阿莫西林噻唑酸,然后用足够的、定量的碘与其发生反应,之后再用准硫代硫酸钠滴定剩余的碘,从而测出消耗碘的阿莫西林的量,最终测出制剂中阿莫西林的含量。
3.电位滴定法电位滴定法是利用阿莫西林的降解产物能够与汞盐(Hg2+)发生反应,形成巯基化物汞盐这一特性而得以测定制剂中阿莫西林含量的,青霉素类化合物的降价降解产物普遍存在这一特性。
该种方法先将阿莫西林制剂水解,然后以铂电极为指示电极、Hg-Hg2SO4为参比电极,然后用硝酸汞滴定液(0.02mol/L)滴定,最终测得制剂中阿莫西林的含量。

阿莫西林检测方法阿莫西林检测方法拼音名:A moxilin英文名:A moxicillin书页号:2000年版二部-338C16H19N3O5S 419.46本品为(2S,5R,6R)-3,3- 二甲基-6-[(R)-(-)-2-氨基-2-(4-羟基苯基)乙酰氨基]-7- 氧代-4-硫杂-1-氮杂双环[3.2.0] 庚烷-2-甲酸三水合物。
按无水物计算,含C16H19N3O5S 不得少于95.0%。
【性状】本品为白色或类白色结晶性粉末;味微苦。
本品在水中微溶,在乙醇中几乎不溶。
比旋度取本品,精密称定,加水溶解并稀释制成每1ml中含1mg 的溶液,依法测定(附录ⅥE),比旋度为+290°至+310°。
【鉴别】(1) 在含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品主峰的保留时间一致。
(5) 本品的红外光吸收图谱应与对照的图谱(光谱集441 图)一致。
【检查】酸度取本品,加水制成每1ml 中含5mg 的溶液,在50℃水浴中微温使溶解后,依法测定(附录ⅥH),pH值应为3.5 ~5.5 。
溶液的澄清度取本品2份,各1.0g,分别加0.5mol/L盐酸溶液10ml及2mol/L氨溶液10ml溶解后立即观察,溶液均应澄清。
如显浑浊,与2号浊度标准液(附录ⅨB)比较,不得更浓。
水分取本品,照水分测定法(附录ⅧM第一法A)测定,含水分应为12.0%~15.0%。
【含量测定】照高效液相色谱法(附录ⅤD)测定。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;以磷酸盐缓冲液(PH5.0)(取磷酸二氢钾13.6g,加水溶解后稀释到2000ml,用8mol/L氢氧化钾溶液调节PH值至5.0±0.1)-乙腈(96:4)为流动相;流速为每分钟约1ml;检测波长为254nm。
理论板数按阿幕西林峰计算应不低于1700。
测定法取本品约30mg,精密称定,置50ml量瓶中,加磷酸盐缓冲液(PH5.0)溶解并稀释至刻度,摇匀,取20μl注入液相色谱仪,记录色谱图;另取阿莫西林对照品适量,同法测定。


阿莫西林聚合物测定原理和几点注意事项阿莫西林,是一种广泛应用于临床的抗生素,能够有效治疗多种感染疾病。
在药学研究和制药过程中,准确测定阿莫西林的聚合物含量十分重要。
本文将介绍阿莫西林聚合物测定的原理以及几点注意事项。
阿莫西林聚合物测定的原理主要基于高效液相色谱(HPLC)技术。
HPLC是一种以溶剂为流动相,将混合物中的化合物通过色谱柱进行分离并进行定量分析的方法。
在阿莫西林聚合物测定中,常常采用反相色谱柱作为分离柱,使用一个特定的流动相体系,在一定的色谱条件下,将样品中的阿莫西林聚合物进行定量测定。
在进行阿莫西林聚合物测定时,需要注意以下几点:1. 样品制备:样品制备是影响测定结果准确性的关键步骤之一。
首先,要确保取样的样品是代表性的,并且与待测样品具有相同的性质。
然后,根据实验要求,精确称取样品,并采用相应的稀释方法将待测样品控制在合适的浓度范围内。
最后,使用适当的提取溶剂将阿莫西林聚合物从样品中提取出来,以便后续的测定分析。
2. 色谱条件设置:在进行阿莫西林聚合物测定时,正确设置色谱条件对于获取准确的结果至关重要。
首先,选择适当的反相色谱柱,根据样品的特性和分离要求确定流动相的组成。
其次,设置恰当的流速和柱温,以确保样品在色谱柱中有足够的停留时间来分离成分。
最后,确定检测波长和进样量,以及其他相关的仪器参数。
3. 内标法测定:为提高测定结果的准确性,常常使用内标法进行阿莫西林聚合物的测定。
内标物是在样品中添加的一种化合物,与待测物具有相同或相似的特性。
通过测定样品中内标物和待测物的峰面积比值,可以消除实验过程中一些因素的影响,提高结果的准确性和可靠性。
4. 定量计算和结果分析:根据测定的峰面积或峰高,结合标定曲线或内标法计算出阿莫西林聚合物的含量。
在计算过程中,需要注意消除样品中可能存在的杂质对结果的干扰。
最后,对测定结果进行统计学分析,并与相关的规范要求进行比较,评估测定的准确性和可靠性。


阿莫西林的质量控制及检测方法引言:阿莫西林是一种广泛应用于临床的抗生素,常用于治疗呼吸道、泌尿道和皮肤软组织感染等疾病。
作为一名专业医学人员,了解阿莫西林的质量控制及检测方法对于保障患者的用药安全至关重要。
本文将深入探讨阿莫西林的质量控制和检测方法,以帮助读者更好地了解这一药物。
一、质量控制的重要性阿莫西林作为一种抗生素,其药物质量直接关系到患者的治疗效果和安全性。
因此,对阿莫西林的质量进行严格控制是至关重要的。
质量控制的目标是确保阿莫西林的纯度、稳定性和安全性,以提供高质量的药物给患者使用。
二、质量控制的方法1. 化学分析方法化学分析方法是评估阿莫西林质量的主要手段之一。
常用的化学分析方法包括高效液相色谱法(HPLC)、气相色谱法(GC)和红外光谱法(IR)等。
这些方法可以通过测定药物的含量、杂质和纯度等指标来评估阿莫西林的质量。
2. 物理分析方法物理分析方法是评估阿莫西林质量的另一种重要手段。
常用的物理分析方法包括粒度分析、溶解度测定和热分析等。
这些方法可以评估阿莫西林的颗粒大小、溶解度和热稳定性等特性,从而判断其质量是否符合标准要求。
3. 生物学分析方法生物学分析方法主要用于评估阿莫西林的生物活性和生物等效性。
常用的生物学分析方法包括细菌抑制试验和动物药效学试验等。
这些方法可以评估阿莫西林的抗菌活性和药效学特性,从而判断其治疗效果和安全性。
三、质量控制的关键指标1. 含量阿莫西林的含量是评估其质量的重要指标之一。
高效液相色谱法(HPLC)是常用的测定阿莫西林含量的方法。
根据药典要求,阿莫西林的含量应在90%~110%之间,以确保其疗效的稳定性和安全性。
2. 杂质阿莫西林的杂质是评估其质量的另一个重要指标。
常见的阿莫西林杂质包括阿莫西林酸、阿莫西林二聚体和阿莫西林酯等。
高效液相色谱法(HPLC)和气相色谱法(GC)是常用的测定阿莫西林杂质的方法。
根据药典要求,阿莫西林的杂质含量应在一定范围内,以确保其纯度和安全性。

阿莫西林色谱条件(依据《中国药典》2015版)色谱柱:Capcell PAK C18;4.6mm 250mm流动相:50 mmol/L (0.05mol/L)磷酸二氢钾溶液(用2mol/L氢氧化钾溶液调节pH值至5.0)-乙腈(97.5:2.5)流速:1.0 mL/min柱温:25℃检测:UV 254nm进样量:20 µL样品浓度:以阿莫西林计0.5mg/mL,溶剂为流动相。
阿莫西林-β-环糊精包合物的制备及包合率测定包合对象:阿莫西林C16H19N3O5S 365.1包合辅料:β-环糊精11351、β-环糊精的纯化:10 gβ-环糊精溶于50 mL去离子水,加热至完全溶解后,趁热过滤,滤液置于冰水中会有环糊精结晶析出,再过滤,得到β-环糊精的晶体,如此重复三次,将得到的β-环糊精干燥备用。
2、线性关系考察-标曲精密称取阿莫西林对照品50.26mg,置于50mL容量瓶中,用流动相制成每0.1 mg/mL 的溶液,摇匀;精密吸取上述对照品溶液1、2、3、4、5 mL,分别置于10 mL容量瓶中,加流动相稀释至刻度,摇匀。
分别进样20 微升,以峰面积(Y)对浓度(C)绘制标准曲线,得到线性方程Y=5.582C+5.278(r=0.9998),可见阿莫西林在0.2001-0.5026mg/mL范围内呈线性关系。
3、阿莫西林包合物制备方法文献一、取β-环糊精加适量蒸馏水,在磁力搅拌器上加热至80℃,溶解后搅拌至室温,分别加入阿莫西林,搅拌2h,制备的包合物溶液置于冰箱中冷藏12h,在抽滤并洗涤,得到沉淀物于60℃干燥。
即得。
文献二、采用超声法制备包合物:称取阿莫西林与β-环糊精饱和水溶液混合后中,超声,40 min 后,得到白色混悬液,抽滤,沉淀依次用蒸馏水快速洗去未包合的阿莫西林和β-环糊精。
60℃干燥得到包合物。
文献三、先将阿莫西林溶解于水溶液中(80度,含2wt%冰醋酸),充分恒温搅拌以使阿莫西林完全溶解。

不同溶剂量下阿莫西林的含量测定实验-药学论文-基础医学论文-医学论文——文章均为WORD文档,下载后可直接编辑使用亦可打印——阿莫西林胶囊适用于敏感菌(不产内酰胺酶菌株)所致的下列感染,溶血链球菌、肺炎链球菌、葡萄球菌或流感嗜血杆菌所致中耳炎、鼻窦炎、咽炎、扁桃体炎等上呼吸道感染;大肠埃希菌、奇异变形杆菌或粪肠球菌所致的泌尿生殖道感染;溶血链球菌、葡萄球菌或大肠埃希菌所致皮肤软组织感染;溶血链球菌、肺炎链球菌、葡萄球菌或流感嗜血杆菌所致急性支气管炎、肺炎等下呼吸道感染;急性单纯性淋病。
本品尚可用于治疗伤寒、伤寒带菌者及钩端螺旋体病;阿莫西林亦可与克拉霉素、兰索拉唑三联用药根除胃、十二指肠幽门螺杆菌,降低消化道溃疡复发率。
阿莫西林胶囊收载在《中国药典》2010 年版二部第403页。
在其检验标准中要求对阿莫西林的含量进行测定,笔者在实验中发现溶解的溶剂量不同,对结果会造成较大的影响,甚至会出现误判。
笔者对几种溶解的溶剂量进行了实验,与大家探讨。
1 试药与仪器1.1 试药:阿莫西林胶囊(石家庄莫药厂批号:130629 标示量0.25g);阿莫西林对照品(中检所批号:130409-201011含量:85.8%)。
1.2 仪器:sartorius cp224s 万分之一天平;sartorius ME235S 十万分之一天平;岛津20A 高效液相色谱仪。
2 方法2.1 色谱条件:以0.05mol/L 磷酸二氢钾溶液(pH 值5.0)∶乙腈(97.5∶2.5)为流动相,流速 1.0ml/min,柱温30.0∶,检查波长254nm,进样量20uL。
2.2 色谱柱:C18柱thermo 5 250mm4.6mm 1005404Q4。
2.3 对照品溶液的制备:取阿莫西林对照品,称量:0.01467g 置25ml量瓶中,用流动相溶解稀释至刻度,即得,浓度为0.5035mg/ml。
2.4 供试液:取样品20 粒,按装量差异检查法称出平均装量0.2980g,其内容物混匀,按照药典规定的取样量取样,共取十二组,每组两个样。

欢迎共阅实验四HPLC法测定阿莫西林胶囊的含量
实验目的: 1. 熟练掌握安捷伦1100液相色谱仪的操作
2. 对液相色谱仪出现的一些故障和维护会进行简单维护
3. 通过外标一点法计算药物浓度
3.实验方法及结果
参照2010年中国药典二部收载阿莫西林胶囊剂的含量测定采用HPLC法(页码
401-404),阿莫西林峰与杂质峰的分离度需大于1.5。
3.1流动相的配制称取6.8 g磷酸二氢钾固体粉末,用1 L纯水溶解;量筒精密量取975 mL磷酸二氢钾水溶液于试剂瓶中,精密量取25 mL乙腈溶液,两相混合,超声10 min后用于流动相;
3.2 对照品溶液的制备精密称取阿莫西林对照品适量, 置50 mL量瓶中,用流动相溶解并稀释至刻度,摇匀,配制成0.5 mg/mL的标准溶液, 备用。
3.3 样品溶液的制备取阿莫西林胶囊5粒,准确称量后取出内容物,研匀。
精密称
)
4.5 所有进样结束后,需对管路进行用色谱甲醇或乙腈进行冲洗,色谱柱也用色谱甲醇或色谱乙腈进行冲洗至少10 min后,保存色谱柱待下次使用。
5. 实验解答题
5.1 采用HPLC-UV法进行含量测定对化合物结构的要求?如何确定紫外最大吸收波长?
5.2 简答采用HPLC法对某一药物进行含量测定的基本思路。
附件:阿莫西林聚合物测定方法的质量标准(草稿)与研究资料阿莫西林聚合物测定法(草稿)阿莫西林聚合物照分子排阻色谱法(中国药典2005年版二部附录ⅤH)测定色谱条件与系统适用性试验用葡聚糖凝胶G-10(40~120μm)为填充剂,玻璃柱(1.0×30cm),流动相A为pH8.0的0.05mol/L磷酸盐缓冲液(取0.05mol/L磷酸氢二钠95ml和0.05mol/L磷酸二氢钠5ml,混合均匀,滤过)。
流动相B为水,流速每分钟1.5ml,检测波长为254nm, 量取0.2mg/ml蓝色葡聚糖2000溶液100μl, 注入液相色谱仪, 分别以流动相A,B进行测定,记录色谱图。
按蓝色葡聚糖2000峰计算理论板数均不低于500,拖尾因子均应小于 2.0。
在两种流动相系统中蓝色葡聚糖2000峰的保留时间比值应在0.93~1.07之间,对照溶液主峰与供试品溶液中聚合物峰与相应色谱系统中蓝色葡聚糖2000峰的保留时间的比值均应在0.93~1.07之间。
称取阿莫西林约0.2g置10ml量瓶中,加2%无水碳酸钠溶液4ml使溶解后,用0.3mg/ml 的蓝色葡聚糖2000溶液稀释至刻度,摇匀。
量取100μl注入液相色谱仪,用流动相A进行测定,记录色谱图。
高聚体的峰高与单体与高聚体之间的谷高比应大于2.0。
另以流动相B为流动相,精密量取对照溶液100μl,连续进样5次,峰面积的相对标准偏差应不大于5.0%。
对照溶液的制备取青霉素对照品适量,精密称定,加水溶解并定量制成每1ml中约含青霉素0.2mg的溶液。
测定法取本品约0.2g, 精密称定,置10ml量瓶中,加2%无水碳酸钠溶液4ml,使溶解后,用水稀释至刻度,摇匀。
立即精密量取100μl注入液相色谱仪,以流动相A为流动相进行测定,记录色谱图。
另精密量取对照溶液100μl注入液相色谱仪,以流动相B为流动相进行测定,记录色谱图。
按外标法以峰面积计算,结果除以10, 即得,含阿莫西林聚合物以阿莫西林计不得过0.15% (阿莫西林: 青霉素=1: 10)。
阿莫西林的分析与检验1阿莫西林化学式化学名为(2S,5R,6R)-3,3-二甲基-6[(R)-(-)-2-氨基-2-(4-羟基苯基)乙酰胺基]-7-氧代-4硫杂-1-氮杂双环[3,2,0]庚烷-2-甲酸散水合物。
又名羟苄西林,是氨苄西林的衍生物。
2药理特性:本品为半合成广谱青霉素,对革兰氏阴性菌如淋球菌,流感杆菌,百日咳杆菌,大肠杆菌,布氏杆菌等作用强,对革兰氏阳性菌的作用与青霉素相同或稍低,其耐酸可口服但不耐酶,可产生抗药性。
3性状:本品为白色或类白色粉末,味微苦,本品在水中微溶,在乙醇中几乎不溶,4采用胶囊剂型的优势4.1阿莫西林味微苦,将阿莫西林密封于胶囊内,可掩盖该药物的苦味,使其外形美观易于携带。
4.2阿莫西林在一定条件的水溶液下不稳定4.2.1可发生降解,引起聚合反应4.2.2水中有磷酸盐,山梨醇,硫酸锌,二乙醇氨等存在时,则会发生分子内成环反应,生成2,5吡嗪二酮。
4.2.3将阿莫西林包入胶囊中,可提高其对水分的稳定性,4.3本品阿莫西林结构中有酚羟基,易发生自动氧化,在光热及重金属催化下,氧化反应加速,将阿莫西林包入胶囊剂,可提高其对光线和空气的稳定性。
5阿莫西林胶囊项下胶囊的检验,5.1外观:胶囊剂应整洁不应有粘连,变形和破损现象,并应无异臭。
5.2装量差异:除另有规定外,应取供试品20粒,分别精密称重后,倾出内容物,(不得损害胶囊壳),用小刷或其它适宜用具拭净,再分别精密称定囊壳重量,求出每粒内容物的装量,每粒的装量与平均的装量相比较,超出装量差异,限度的胶囊,不得多于2粒,并不得有一粒超出限度值的一倍。
平均装量为0.3克以下的,装量差异限度为±10%,平均装量为,0.3g或0.3g以上的,装量差异限度为±7.5%。
5.3崩解时限的检查崩解时限的检查,采用生降式崩解仪,其主要结构为一能升降的金属支架与下端镶有筛网的吊篮,并附有挡板。
测定时使用胶囊剂在液体介质中,若胶囊剂漂浮在页面,可加挡板,阿莫西林胶囊剂应在30min内崩解,如有一粒不能完全崩解,则另取6粒重复试验,均应符合规定。
阿莫西林
色谱条件(依据《中国药典》2015版)
色谱柱:Capcell PAK C18;4.6mm 250mm
流动相:50 mmol/L (0.05mol/L)磷酸二氢钾溶液(用2mol/L氢氧化钾溶液调节pH值至5.0)-乙腈(97.5:2.5)
流速:1.0 mL/min
柱温:25℃
检测:UV 254nm
进样量:20 µL
样品浓度:以阿莫西林计0.5mg/mL,溶剂为流动相。
阿莫西林-β-环糊精包合物的制备及包合率测定
包合对象:阿莫西林C16H19N3O5S 365.1
包合辅料:β-环糊精1135
1、β-环糊精的纯化:10 gβ-环糊精溶于50 mL去离子水,加热至完全溶解后,趁热过滤,滤液置于冰水中会有环糊精结晶析出,再过滤,得到β-环糊精的晶体,如此重复三次,将
得到的β-环糊精干燥备用。
2、线性关系考察-标曲
精密称取阿莫西林对照品50.26mg,置于50mL容量瓶中,用流动相制成每0.1 mg/mL 的溶液,摇匀;精密吸取上述对照品溶液1、2、3、4、5 mL,分别置于10 mL容量瓶中,加流动相稀释至刻度,摇匀。
分别进样20 微升,以峰面积(Y)对浓度(C)绘制标准曲线,得到线性方程Y=5.582C+5.278(r=0.9998),可见阿莫西林在0.2001-0.5026mg/mL范围内呈线性关系。
3、阿莫西林包合物制备方法
文献一、取β-环糊精加适量蒸馏水,在磁力搅拌器上加热至80℃,溶解后搅拌至室温,分别加入阿莫西林,搅拌2h,制备的包合物溶液置于冰箱中冷藏12h,在抽滤并洗涤,得到沉淀物于60℃干燥。
即得。
文献二、采用超声法制备包合物:
称取阿莫西林与β-环糊精饱和水溶液混合后中,超声,40 min 后,得到白色混悬液,抽滤,沉淀依次用蒸馏水快速洗去未包合的阿莫西林和β-环糊精。
60℃干燥得到包合物。
文献三、先将阿莫西林溶解于水溶液中(80度,含2wt%冰醋酸),充分恒温搅拌以使阿莫
西林完全溶解。
然后,环糊精缓慢加入上述溶液中,混合物继续维持在80度搅拌1h,过滤,
溶剂用旋蒸法除去,在用真空泵抽干,得到白色粉末,空气中自然晾干。
4、包合物的鉴定:(紫外、热分析、红外、XRD、薄层色谱)
分别对包合物溶液,阿莫西林溶液,环糊精与阿莫西林的混合液以及环糊精的水溶液4种样品进行紫外扫面观察峰的位置变化。
薄层色谱法(展开剂=乙酸乙酯-丙酮-冰醋酸-水(5:2:2:1))
5、包合物中阿莫西林的含量测定:
称取50mg包合物,用水溶解,超声10min,过滤,滤液测定其浓度。
计算包合率。
包合率=测得阿莫西林质量/投的阿莫西林质量×100%
收率=包合物质量/阿莫西林质量+环糊精质量×100%
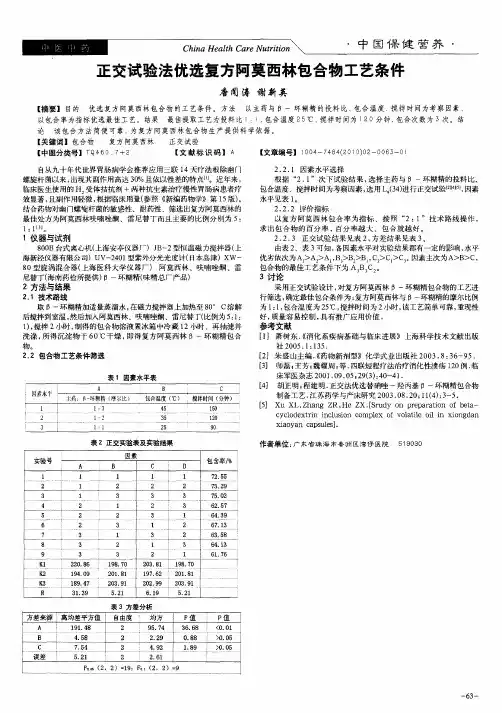
正交试验设计
表三因素三水平表
水平A:投料比(mol)B:时间(h)C:温度(℃)
1 1:1
2 25
2 1:2
3 35
3 1:3
4 45
试验次数9
表正交实验表及实验结果
实验号 A B C D 包合率/%
1 1 1 1 1
2 1 2 2 2
3 1 3 3 3
4 2 1 2 3
5 2 2 3 1
6 2 3 1 2
7 3 1 3 2
8 3 2 1 3
9 3 3 2 1
K1
K2
K3
R。