阿莫西林聚合物测定原理和几点注意事项
- 格式:doc
- 大小:21.50 KB
- 文档页数:3

实验十二阿莫西林胶囊的分析一、实验目的1.熟悉高效液相色谱仪的结构及正确使用。
2.掌握阿莫西林胶囊分析的原理及方法。
二、实验原理阿莫西林为β–内酰胺类抗生素,其母核部分与侧链部分均有共轭体系,有紫外吸收,因此阿莫西林胶囊的鉴别、有关物质的检查、含量测定均采用反相高效液相色谱法。
有关物质的检查方法为外标法和不加校正因子的主成分自身对照法,含量测定采用外标法定量。
三、实验方法本品含C16H19 N3O5S应为标示量的90.0%~110.0%。
【鉴别】在含量测定项下记录的色谱图中,供试品溶液主峰的保留时问应与对照品溶液主峰的保留时间一致。
【检查】水分取本品的内容物,照水分测定法(附录M第一法A)测定,含水分不得过16.0%。
有关物质取本品的内容物适量,精密称定,用流动相A溶解并制成每1ml中含2mg 的溶液,滤过,取续滤液照阿莫西林有关物质项下的方法测定。
单个杂质的峰面积不得大于对照溶液主峰面积的2倍(2.0%),各杂质峰面积的和不得大于对照溶液主峰面积的5倍(5.0%)。
供试品溶液中汪何小于对照溶液主峰面积0.05倍的峰可忽略不计)溶出度取本品,照溶出度测定法(附录X C第一法),以水900ml为溶出介质,转速为每分钟100转,依法操作,经45分钟时,取溶液适量,滤过,精密量取续滤液适量,用溶出介质稀释成缈"ml中约含130μg的溶液,照紫外一可见分光光度法(附录ⅣA),在272nm 的波长处测定吸光度;另取装量差异项下的内容物,混合均匀,精密称取适量(约相当于平均装量),按标示量加溶出介质溶解并稀释成每1ml中约含130μg的溶液,滤过,取续滤液作为对照溶液,同法测定,计算每粒的溶出量。
限度为80%,应符合规定。
其他应符合胶囊片剂项下有关的各项规定(附录I A)。
【含量测定】1.色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;以0.05mol/L磷酸二氢钾溶液(2mol/L氢氧化钠溶液调节PH值至5.0)–乙(97.5︰2.5)为流动相;流速为1ml/min,检测波长为254nm,理论板数按阿莫西林峰计算不低于2000。
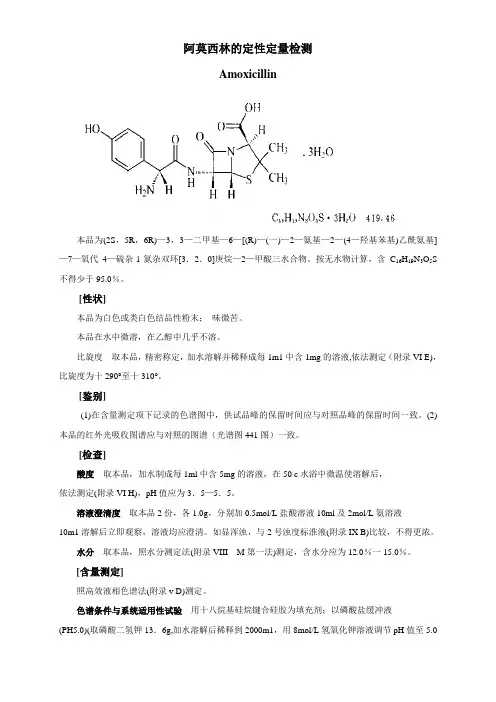
阿莫西林的定性定量检测Amoxicillin本品为(2S,5R,6R)—3,3—二甲基—6—[(R)—(一)—2—氨基—2—(4—羟基苯基)乙酰氨基]—7—氧代4—硫杂-1-氮杂双环[3.2.0]庚烷—2—甲酸三水合物。
按无水物计算,含C16H19N3O5S 不得少于95.0%。
[性状]本品为白色或类白色结品性粉末;味微苦。
本品在水中微溶,在乙醇中几乎不溶。
比旋度取本品,精密称定,加水溶解并稀释成每1m1中含1mg的溶液,依法测定(附录VI E),比旋度为十290°至十310°。
[鉴别](1)在含量测定项下记录的色谱图中,供试品峰的保留时间应与对照品峰的保留时间一致。
(2)本品的红外光吸收图谱应与对照的图谱(光谱图441图)一致。
[检查]酸度取本品,加水制成每1ml中含5mg的溶液,在50 c水浴中微温使溶解后,依法测定(附录VI H),pH值应为3.5—5.5。
溶液澄清度取本品2份,各1.0g,分别加0.5mol/L盐酸溶液10ml及2mol/L氨溶液10m1溶解后立即观察,溶液均应澄清。
如显浑浊,与2号浊度标淮液(附录IX B)比较,不得更浓。
水分取本品,照水分测定法(附录VIII M第一法)测定,含水分应为12.0%一15.0%。
[含量测定]照高效液相色谱法(附录v D)测定。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;以磷酸盐缓冲液(PH5.0)(取磷酸二氢钾13.6g,加水溶解后稀释到2000m1,用8mol/L氢氧化钾溶液调节pH值至5.0土0.1)—乙腈(96:4)为流动相;流速为每分钟约1m1;检测波长为254nm。
理论板数按阿莫西林峰计算应不低于1700。
对照品溶液的制备取阿莫西林对照品约60mg,精密称定,置50m1量瓶中,加磷酸盐缓冲液(pH5.0)溶解并稀释至刻度,摇匀。
供试品溶液的制备与测定取本品约120mg,精密称定,置100m1量瓶中,加磷酸盐缓冲液(pH5.0)溶解并稀释至刻度,摇匀,精密量取10µl,注入液相色谱仪,记录色谱图;另取对照品溶液,同法测定。
GMP管理文件一、目的:建立阿莫西林检验的标准操作规程,保证正确操作。
二、依据:《中国兽药典》二00五版一部。
三、适用范围:适用于阿莫西林的检验。
四、责任者:QC检验员五、正文:1.质量标准:(见阿莫西林质量标准)。
2.试剂:2.01 0.2mol/L盐酸 2.02 2mol/L氨溶液2.03 0.05mol/L磷酸盐缓冲液 2.04 乙腈2.05 磷酸盐缓冲液(pH8.0) 2.06 2%碳酸钠溶液2.07 0.05mol/L磷酸二氢钾3.仪器与用具3.01 酸度计 3.02 自动水分测定仪3.03 电子天平 3.04 旋光仪3.05 高效液相色谱仪4.操作步骤:4.1 性状本品为白色或类白色结晶性粉末;味微苦。
本品在水中微溶,在乙醇中几乎不溶。
则判定该项合格。
4.1.1 比旋度取本品,精密称定,加水溶解并定量稀释制成每 1ml中含1mg 的溶液,照旋光度测定法(详见旋光度测定法标准操作规程)测定,比旋度为+290°至+310°则判定该项合格。
4.2 鉴别:(1)在含量项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品溶液主峰的保留时间一致。
则判定该项合格。
(2)本品的红外光吸收图谱应与对照的图谱一致。
(委托检验)4.3 检查4.3.1 酸度取本品,加水制成每1ml中含5mg的溶液,在50℃水浴中微温使溶解后,照PH值测定法测定(详见pH值测定法标准操作规程)。
4.3.2 溶液的澄清度取本品5份,各1.0g,分别加0.5mol/L盐酸溶液10ml 及2mol/L氨溶液10ml溶解后立即观察,溶液均应澄清。
如显浑浊,与2号浊度标准液(详见溶液澄清度检查法标准操作规程)比较,均不得更浓。
则判定该项合格。
4.3.3 有关物质取本品适量,精密称定,加流动相A溶解并稀释成每1ml中含2mg的溶液,作为供试品溶液;另取阿莫西林对照品适量,精密称定,加流动相A 溶解并稀释成每1ml中含20μg的溶液作为对照溶液。

实验十二阿莫西林胶囊的分析AmoxilinA moxiciHinCjiHijNjOjS * SHrO 419*46一、实验目的1熟悉高效液相色谱仪的结构及正确使用。
2 •掌握阿莫西林胶囊分析的原理及方法。
二、实验原理阿莫西林为B-内酰胺类抗生素,其母核部分与侧链部分均有共轭体系,有紫外吸收, 因此阿莫西林胶囊的鉴别、有关物质的检查、含量测定均采用反相高效液相色谱法。
有关 物质的检查方法为外标法和不加校正因子的主成分自身对照法,含量测定采用外标法定量。
三、实验方法本品含 C 16H 19 N 3O 5S 应为标示量的 90.0% ~110.0%。
【鉴别】 在含量测定项下记录的色谱图中,供试品溶液主峰的保留时问应与对照品溶 液主峰的保留时间一致。
【检查】 水分 取本品的内容物,照水分测定法 (附录M 第一法A )测定,含水分不得 过 16.0%。
有关物质取本品的内容物适量, 精密称定,用流动相A 溶解并制成每1ml 中含2mg 的溶液,滤过,取续滤液照阿莫西林有关物质项下的方法测定。
单个杂质的峰面积不得大 于对照溶液主峰面积的2倍(2.0 % ),各杂质峰面积的和不得大于对照溶液主峰面积的 5倍 (5.0% )。
供试品溶液中汪何小于对照溶液主峰面积 0.05倍的峰可忽略不计) 溶出度 取本品,照溶出度测定法(附录X C 第一法),以水900ml 为溶出介质,转速 为每分钟100转,依法操作,经 45分钟时,取溶液适量,滤过,精密量取续滤液适量,用 溶出介质稀释成缈"ml 中约含130⑷的溶液,照紫外一可见分光光度法(附录W A ),在272nm 的波长处测定吸光度;另取装量差异项下的内容物,混合均匀,精密称取适量(约相当于平 均装量),按标示量加溶出介质溶解并稀释成每1ml 中约含130⑷的溶液,滤过,取续滤液 作为对照溶液,同法测定,计算每粒的溶出量。
限度为80%,应符合规定。
其他 应符合胶囊片剂项下有关的各项规定(附录I A )。

阿莫西林聚合物测定原理和几点注意事项阿莫西林,是一种广泛应用于临床的抗生素,能够有效治疗多种感染疾病。
在药学研究和制药过程中,准确测定阿莫西林的聚合物含量十分重要。
本文将介绍阿莫西林聚合物测定的原理以及几点注意事项。
阿莫西林聚合物测定的原理主要基于高效液相色谱(HPLC)技术。
HPLC是一种以溶剂为流动相,将混合物中的化合物通过色谱柱进行分离并进行定量分析的方法。
在阿莫西林聚合物测定中,常常采用反相色谱柱作为分离柱,使用一个特定的流动相体系,在一定的色谱条件下,将样品中的阿莫西林聚合物进行定量测定。
在进行阿莫西林聚合物测定时,需要注意以下几点:1. 样品制备:样品制备是影响测定结果准确性的关键步骤之一。
首先,要确保取样的样品是代表性的,并且与待测样品具有相同的性质。
然后,根据实验要求,精确称取样品,并采用相应的稀释方法将待测样品控制在合适的浓度范围内。
最后,使用适当的提取溶剂将阿莫西林聚合物从样品中提取出来,以便后续的测定分析。
2. 色谱条件设置:在进行阿莫西林聚合物测定时,正确设置色谱条件对于获取准确的结果至关重要。
首先,选择适当的反相色谱柱,根据样品的特性和分离要求确定流动相的组成。
其次,设置恰当的流速和柱温,以确保样品在色谱柱中有足够的停留时间来分离成分。
最后,确定检测波长和进样量,以及其他相关的仪器参数。
3. 内标法测定:为提高测定结果的准确性,常常使用内标法进行阿莫西林聚合物的测定。
内标物是在样品中添加的一种化合物,与待测物具有相同或相似的特性。
通过测定样品中内标物和待测物的峰面积比值,可以消除实验过程中一些因素的影响,提高结果的准确性和可靠性。
4. 定量计算和结果分析:根据测定的峰面积或峰高,结合标定曲线或内标法计算出阿莫西林聚合物的含量。
在计算过程中,需要注意消除样品中可能存在的杂质对结果的干扰。
最后,对测定结果进行统计学分析,并与相关的规范要求进行比较,评估测定的准确性和可靠性。

实验四 HPLC法测定阿莫西林胶囊的含量实验目的: 1. 熟练掌握安捷伦1100液相色谱仪的操作2. 对液相色谱仪出现的一些故障和维护会进行简单维护3. 通过外标一点法计算药物浓度4. 采用高效液相色谱法对阿莫西林胶囊主成分阿莫西林进行含量测定实验原理:利用高效液相色谱仪的高选择性、高灵敏度等特点,通过记录已知浓度的阿莫西林对照品的色谱峰面积,并测定未知浓度的阿莫西林胶囊的色谱峰面积,根据外标一点法计算胶囊中阿莫西林的标示量的百分含量。
实验背景:阿莫西林 (Amoxicillin) 为β-酰胺类抗生素, 临床疗效好, 可口服且不易引起过敏。
2010年中国药典二部收载阿莫西林胶囊剂的含量测定采用HPLC法(页码401-404),方法操作简便、快速、准确、专属性好。
1.仪器与试药Agilent 1100高效液相色谱仪: G1310 泵, G2280进样器, G1314A紫外波长检测器。
阿莫西林对照品由中国食品药品检定研究院提供(纯度为86.4%), 乙腈为色谱纯, 磷酸二氢钾为分析纯,阿莫西林胶囊由国某药厂提供。
50 ml容量瓶,小镊子;棉花;无水乙醇2.色谱条件色谱柱: Zorbax SB-C色谱柱(4.6 mm × 150 mm, 5 µm) ; 流动相: 50 mmol/L18磷酸二氢钾溶液:乙腈 (97.5 : 2.5, v/v), 流速1.0 ml/min ; 检测波长254 nm,进样量20 μl,柱温室温。
3.实验方法及结果参照2010年中国药典二部收载阿莫西林胶囊剂的含量测定采用HPLC法(页码401-404),阿莫西林峰与杂质峰的分离度需大于1.5。
3.1流动相的配制称取6.8 g磷酸二氢钾固体粉末,用1 L纯水溶解;量筒精密量取975 mL磷酸二氢钾水溶液于试剂瓶中,精密量取25 mL乙腈溶液,两相混合,超声10 min后用于流动相;3.2 对照品溶液的制备精密称取阿莫西林对照品适量, 置50 mL量瓶中,用流动相溶解并稀释至刻度,摇匀,配制成0.5 mg/mL的标准溶液, 备用。


阿莫西林胶囊中高分子杂质含量的凝胶色谱法测定作者:贺亮张倩来源:《科学与财富》2015年第35期摘要:本文对阿莫西林胶囊中高分子杂质的含量测定方法进行研究。
固定相为:葡聚糖凝胶G-10(40-120um)为填充剂,玻璃柱内径1.0-1.4cm,柱长30-40cm;pH值为8.0的0.05mol/L磷酸盐缓冲溶液[0.05mol/L磷酸氢二钠-0.05mol/L磷酸二氢钠(95:5)]为A流动相,水为B流动相;检测波长为254nm;流速1.5mL·min-1,柱温:30℃。
此方法简便、准确、重现性好,可用于阿莫西林胶囊中高分子杂质的含量测定。
关键词:阿莫西林胶囊;高分子杂质;含量阿莫西林,又名安莫西林或安默西林,是一种最常用的半合成青霉素类广谱β-内酰胺类抗生素。
阿莫西林杀菌作用强,穿透细胞膜的能力也强。
在临床上最常见的不良反应是速发型过敏反应。
引发β-内酰胺抗生素速发型过敏反应的过敏原并不是β-内酰胺抗生素本身,而是其中存在的高分子聚合物。
高分子聚合物具有强的抗原性质,在临床过敏反应中起重要作用。
高分子杂质:高分子杂质是对药品中分子量大于药物本身的杂质总称。
临床应用阿莫西林的不良反应发生率约为5-6%,其主要不良反应有:药物热、荨麻疹、皮疹、哮喘、过敏性休克、白细胞减少、血小板减少、腹泻、恶心、呕吐、渗出性多形性红斑、Lyell综合征、肝功能紊乱、肾功能紊乱、剥脱性皮炎、焦虑、失眠、头晕以及行为异常等中枢神经系统症状等[1-6]。
凝胶色谱法又叫凝胶色谱技术,是六十年代初发展起来的一种快速而又简单的分离分析技术,由于设备简单、操作方便,不需要有机溶剂,对高分子物质有很高的分离效果。
凝胶色谱法主要用于高聚物的相对分子质量分级分析以及相对分子质量分布测试。
本文采用高校液相测定了阿莫西林胶囊中高分子杂质的含量。
1 仪器与试药6000LDI高效液相色谱仪(天津琛航科苑科技发展有限公司);UV1902双光束紫外可见分光光度计(上海棱光技术有限公司);赛多利斯Cubis?高端天平(德国赛多利斯集团);OHAUS 奥豪斯 STARTER 3100 PH计(上海泉佰仪器有限公司);ULABO 实验室温度控制器(优莱博技术(北京)有限公司);JULABO TW系列通用水浴槽(优莱博技术(北京)有限公司)。

阿司匹林含量测定原理阿司匹林是一种常用的药物,它具有镇痛、退烧、抗炎等功效,因此被广泛应用于临床。
然而,药物的含量对于药效的发挥具有至关重要的作用,因此需要对药物中的含量进行准确的测定。
本文将介绍阿司匹林含量测定的原理及相关方法。
首先,阿司匹林的含量测定是通过化学分析方法来进行的。
常用的方法包括酸碱滴定法、光度法、高效液相色谱法等。
其中,酸碱滴定法是一种简单而常用的方法。
其原理是将阿司匹林溶解在酸性介质中,然后用碱溶液滴定至中和终点,通过滴定液的用量计算出阿司匹林的含量。
光度法则是利用阿司匹林在特定波长下的吸光度与其浓度成正比的原理,通过测定吸光度来计算阿司匹林的含量。
而高效液相色谱法则是通过样品在色谱柱中的分离和检测来测定阿司匹林的含量。
其次,阿司匹林含量测定的原理是基于化学反应的定量分析。
在酸碱滴定法中,阿司匹林与碱溶液发生中和反应,终点时滴定液的用量与阿司匹林的含量成正比。
而光度法则是利用阿司匹林在特定波长下的吸光度与其浓度成正比的原理,通过比较样品的吸光度与标准曲线来计算出含量。
高效液相色谱法则是通过样品在色谱柱中的分离和检测来测定阿司匹林的含量,其原理是利用不同成分在色谱柱中的分离和检测来进行定量分析。
最后,阿司匹林含量测定的原理是基于化学分析方法的定量分析。
不同的测定方法有其各自的特点和适用范围,选择合适的方法进行测定对于准确测定阿司匹林的含量是非常重要的。
在实际操作中,需要严格按照方法要求操作,保证测定结果的准确性和可靠性。
总之,阿司匹林含量测定是通过化学分析方法进行的定量分析,主要包括酸碱滴定法、光度法、高效液相色谱法等。
这些方法都是基于化学反应的定量分析原理进行的,选择合适的方法进行测定对于准确测定阿司匹林的含量至关重要。
希望本文能够对阿司匹林含量测定的原理有所帮助。

阿司匹林含量测定的方法和原理阿司匹林(aspirin),也称为乙酰水杨酸(acetylsalicylic acid),是一种广泛应用的非处方药和治疗心脑血管疾病的药物。
正确的阿司匹林含量可确保其药效和安全性。
本篇文章将介绍阿司匹林含量测定的方法和原理。
阿司匹林含量测定的方法1.紫外光分光光度法紫外光分光光度法是阿司匹林含量测定的最常用方法。
该方法在特定波长下使用分光光度法测定阿司匹林的吸光度,并使用标准曲线来计算其含量。
标准曲线通常以已知浓度的阿司匹林溶液制备。
该方法的优点包括操作简单、快速和准确。
但是,该方法对样品的制备要求高,且有一定的干扰影响。
2.高效液相色谱法高效液相色谱法也称为HPLC法,是一种基于分子分离的测定方法。
该方法通过样品在色谱柱中的分离,测量阿司匹林成分的相对含量。
HPLC法具有高速、准确、精确和可重复性强的优点,但是需要专业实验技能和复杂的设备。
3.比色法比色法是一种基于颜色反应的测定方法。
阿司匹林分解产物在一定的条件下,会产生梅威氏试剂反应,颜色会发生变化。
该方法适用于测定微量和痕量阿司匹林含量,但是准确度和精度较低。
阿司匹林含量测定的原理阿司匹林含量测定的原理基于化学反应和物理原理。
1.紫外光分光光度法该方法使用的原理是阿司匹林分子在特定波长下的吸光度,通过比较标准和待测样品的吸光度来计算其含量。
阿司匹林在波长为240 nm处,有一个最大吸光度。
该波长下,阿司匹林的分子能够吸收波长为240 nm的紫外线,使分光光度计读数增加。
2.高效液相色谱法该方法利用分子分离原理,通过样品在色谱柱中的分离和检测,测量阿司匹林成分的相对含量。
通过调整柱温、流速和移动相成分,从而实现对分子的分离和检测。
3.比色法该方法利用阿司匹林与梅威氏试剂的化学反应,形成一种明显的带紫色的复合物。
该复合物在一定波长下,具有最大的吸光度,通过比较标准和待测样品的吸光度来测定阿司匹林的含量。
总结阿司匹林含量测定是保证阿司匹林药效和安全性的必要步骤。
![阿莫西林聚合物测定法[1]](https://uimg.taocdn.com/1acb280a0740be1e650e9ad7.webp)
附件:阿莫西林聚合物测定方法的质量标准(草稿)与研究资料阿莫西林聚合物测定法(草稿)阿莫西林聚合物照分子排阻色谱法(中国药典2005年版二部附录ⅤH)测定色谱条件与系统适用性试验用葡聚糖凝胶G-10(40~120μm)为填充剂,玻璃柱(1.0×30cm),流动相A为pH8.0的0.05mol/L磷酸盐缓冲液(取0.05mol/L磷酸氢二钠95ml和0.05mol/L磷酸二氢钠5ml,混合均匀,滤过)。
流动相B为水,流速每分钟1.5ml,检测波长为254nm, 量取0.2mg/ml蓝色葡聚糖2000溶液100μl, 注入液相色谱仪, 分别以流动相A,B进行测定,记录色谱图。
按蓝色葡聚糖2000峰计算理论板数均不低于500,拖尾因子均应小于 2.0。
在两种流动相系统中蓝色葡聚糖2000峰的保留时间比值应在0.93~1.07之间,对照溶液主峰与供试品溶液中聚合物峰与相应色谱系统中蓝色葡聚糖2000峰的保留时间的比值均应在0.93~1.07之间。
称取阿莫西林约0.2g置10ml量瓶中,加2%无水碳酸钠溶液4ml使溶解后,用0.3mg/ml 的蓝色葡聚糖2000溶液稀释至刻度,摇匀。
量取100μl注入液相色谱仪,用流动相A进行测定,记录色谱图。
高聚体的峰高与单体与高聚体之间的谷高比应大于2.0。
另以流动相B为流动相,精密量取对照溶液100μl,连续进样5次,峰面积的相对标准偏差应不大于5.0%。
对照溶液的制备取青霉素对照品适量,精密称定,加水溶解并定量制成每1ml中约含青霉素0.2mg的溶液。
测定法取本品约0.2g, 精密称定,置10ml量瓶中,加2%无水碳酸钠溶液4ml,使溶解后,用水稀释至刻度,摇匀。
立即精密量取100μl注入液相色谱仪,以流动相A为流动相进行测定,记录色谱图。
另精密量取对照溶液100μl注入液相色谱仪,以流动相B为流动相进行测定,记录色谱图。
按外标法以峰面积计算,结果除以10, 即得,含阿莫西林聚合物以阿莫西林计不得过0.15% (阿莫西林: 青霉素=1: 10)。
阿莫西林聚合物测定原理和几点注意事项浙江省食品药品检验所抗生素中的高分子聚合物杂质是引发抗生素药物过敏反应的真正过敏原,因此严格控制抗生素药物中高分子聚合物的含量有着重要的意义。
近日监督抽样中发现,阿莫西林胶囊聚合物含量超限问题较严重,因此我们以阿莫西林胶囊为例,对阿莫西林高分子聚合物测定的原理和测定过程中需要注意的问题进行了研究,现总结如下:一、测定原理和其他β-内酰胺类抗生素的聚合物测定方法一样,阿莫西林胶囊采用Sephadex G-10凝胶色谱系统测定。
Sephadex G-10的排阻分子量为700d,因此除部分寡聚物外,阿莫西林胶囊聚合物在色谱过程中均不保留,即所有的高分子杂质表现为单一的色谱峰,阿莫西林主成分表现为另一色谱峰。
由于高分子聚合物的对照品难以制备,而此利用药品自身对照品在水溶剂及纯水流动相中主成分缔合物作对照,取代高分子杂质对照品对阿莫西林胶囊聚合物进行检测,较好的解决了这一难题;而在缓冲液作为溶剂及流动相的情况下,阿莫西林相对不易聚合,因而在此条件下测定样品中聚合物的含量。
二、几个注意点1、对照品。
进口药品注册标准(JX20000355)和中国药典2005年版二部采用不同的对照品,结果按照进口药品注册标准检测得聚合物含量约为按药典方法的2倍。
差别如此大的主要原因是进口药品注册标准以阿莫西林为对照品,中国药典2005年版二部选用青霉素作为对照品,两种对照品紫外响应不同,因而作为外标计算同一样品中聚合物含量所得结果也不同。
2、溶解方法。
样品溶解可采用加2%Na2CO3溶液助溶和超声溶解两种方法,两种方法所得结果基本上没有差异。
需要注意的是超声溶解时间不宜过长,超声用水不宜过热,否则容易使聚合物含量上升。
b)样品溶解至进样放置时间不宜过长。
C)阿莫西林对照溶液不稳定,在进样过程中峰面积越来越小,峰面积逐渐降低,峰宽逐渐增宽,造成连续进样的峰面积相对标准偏差较大,因此对照品溶解后要马上连续进样,有条件的要使用低温进样保温器。
附件:阿莫西林聚合物测定方法的质量标准(草稿)与研究资料阿莫西林聚合物测定法(草稿)阿莫西林聚合物照分子排阻色谱法(中国药典2005年版二部附录ⅤH)测定色谱条件与系统适用性试验用葡聚糖凝胶G-10(40~120μm)为填充剂,玻璃柱(1.0×30cm),流动相A为pH8.0的0.05mol/L磷酸盐缓冲液(取0.05mol/L磷酸氢二钠95ml和0.05mol/L磷酸二氢钠5ml,混合均匀,滤过)。
流动相B为水,流速每分钟1.5ml,检测波长为254nm, 量取0.2mg/ml蓝色葡聚糖2000溶液100μl, 注入液相色谱仪, 分别以流动相A,B进行测定,记录色谱图。
按蓝色葡聚糖2000峰计算理论板数均不低于500,拖尾因子均应小于 2.0。
在两种流动相系统中蓝色葡聚糖2000峰的保留时间比值应在0.93~1.07之间,对照溶液主峰与供试品溶液中聚合物峰与相应色谱系统中蓝色葡聚糖2000峰的保留时间的比值均应在0.93~1.07之间。
称取阿莫西林约0.2g置10ml量瓶中,加2%无水碳酸钠溶液4ml使溶解后,用0.3mg/ml 的蓝色葡聚糖2000溶液稀释至刻度,摇匀。
量取100μl注入液相色谱仪,用流动相A进行测定,记录色谱图。
高聚体的峰高与单体与高聚体之间的谷高比应大于2.0。
另以流动相B为流动相,精密量取对照溶液100μl,连续进样5次,峰面积的相对标准偏差应不大于5.0%。
对照溶液的制备取青霉素对照品适量,精密称定,加水溶解并定量制成每1ml中约含青霉素0.2mg的溶液。
测定法取本品约0.2g, 精密称定,置10ml量瓶中,加2%无水碳酸钠溶液4ml,使溶解后,用水稀释至刻度,摇匀。
立即精密量取100μl注入液相色谱仪,以流动相A为流动相进行测定,记录色谱图。
另精密量取对照溶液100μl注入液相色谱仪,以流动相B为流动相进行测定,记录色谱图。
按外标法以峰面积计算,结果除以10, 即得,含阿莫西林聚合物以阿莫西林计不得过0.15% (阿莫西林: 青霉素=1: 10)。
阿司匹林片的分析实验注意事项阿司匹林片的分析实验注意事项可以从实验前、实验中和实验后三个方面来详细阐述。
以下是一个可能的1200字以上的回答:实验前:1. 实验室准备:确保实验室具备进行药品分析的必要设备和试剂,如分析天平、移液器、试管、试剂瓶等。
2. 安全措施:阅读并遵守实验室安全操作规程,佩戴个人防护装备,如实验手套、护目镜等。
确保实验室通风良好,并遵守药物分析实验的处理废弃物的相关规定。
3. 试剂的准备:准备好所需的试剂,包括阿司匹林片和用于分析的溶剂等。
实验中:1. 试样的处理:首先,将阿司匹林片样品称取适量,并尽量避免手动处理,以防止样品污染。
可以通过粉碎、溶解等方法,将样品转化为所需的形式。
2. 样品的质量控制:确保样品经过适当的保存和保管,以避免样品质量受损。
在进行分析之前,可以进行适当的质量控制实验,如样品的纯度检验等。
3. 实验参数的设定:根据实验目的和方法要求,设置合适的实验参数,例如pH、温度、反应时间等。
并进行预实验,调整实验条件,以保证实验的准确性和重复性。
4. 仪器操作:熟悉并正确操作所需的仪器设备,如分析天平、试管、电子天平等,确保实验操作的准确性。
5. 实验步骤的认真记录:对实验中所有的步骤和测量结果进行详细记录,包括试剂使用量、待测液的颜色变化、反应时间等,以便后续的数据处理和分析。
实验后:1. 结果的分析与处理:根据实验目的,对实验结果进行分析和处理,包括计算、求平均值、绘制曲线等。
2. 结果的验证:对实验结果进行验证,如使用不同的方法、仪器或进行复查,确保结果的准确性和可靠性。
3. 结果的解释与报告:对实验结果进行解释,并由实验者撰写实验报告。
在报告中包括实验的目的、原理、实验步骤、结果分析和结论等内容。
4. 仪器设备的清洁与维护:保持使用的仪器设备的清洁,并定期进行维护保养,以延长其使用寿命,并保证实验结果的准确性。
5. 废弃物的处理:根据实验室的规定和相关法规,将实验中产生的废弃物分类收集和处理。
阿莫西林聚合物测定原理和几点注意事项
浙江省食品药品检验所
抗生素中的高分子聚合物杂质是引发抗生素药物过敏反应的真正过敏原,因此严格控制抗生素药物中高分子聚合物的含量有着重要的意义。
近日监督抽样中发现,阿莫西林胶囊聚合物含量超限问题较严重,因此我们以阿莫西林胶囊为例,对阿莫西林高分子聚合物测定的原理和测定过程中需要注意的问题进行了研究,现总结如下:
一、测定原理
和其他β-内酰胺类抗生素的聚合物测定方法一样,阿莫西林胶囊采用Sephadex G-10凝胶色谱系统测定。
Sephadex G-10的排阻分子量为700d,因此除部分寡聚物外,阿莫西林胶囊聚合物在色谱过程中均不保留,即所有的高分子杂质表现为单一的色谱峰,阿莫西林主成分表现为另一色谱峰。
由于高分子聚合物的对照品难以制备,而此利用药品自身对照品在水溶剂及纯水流动相中主成分缔合物作对照,取代高分子杂质对照品对阿莫西林胶囊聚合物进行检测,较好的解决了这一难题;而在缓冲液作为溶剂及流动相的情况下,阿莫西林
相对不易聚合,因而在此条件下测定样品中聚合物的含量。
二、几个注意点
1、对照品。
进口药品注册标准(JX20000355)和中国药典2005年版二部采用不同的对照品,结果按照进口药品注册标准检测得聚合物含量约为按药典方法的2倍。
差别如此大的主要原因是进口药品注册标准以阿莫西林为对照品,中国药典2005年版二部选用青霉素作为对照品,两种对照品紫外响应不同,因而作为外标计算同一样品中聚合物含量所得结果也不同。
2、溶解方法。
样品溶解可采用加2%Na2CO3溶液助溶和超声溶解两种方法,两种方法所得结果基本上没有差异。
需要注意的是超声溶解时间不宜过长,超声用水不宜过热,否则容易使聚合物含量上升。
b)样品溶解至进样放置时间不宜过长。
C)阿莫西林对照溶液不稳定,在进样过程中峰面积越来越小,峰面积逐渐降低,峰宽逐渐增宽,造成连续进样的峰面积相对标准偏差较大,因此对照品溶解后要马上连续进样,有条件的要使用低温进样保温器。
3、Sephadex G-10色谱柱的活化。
色谱柱对样品有一定吸附
作用,如有需要可对填料进行活化。
在使用含0.2mol/L氢氧化钠和0.5mol/L氯化钠的溶液冲洗凝胶柱各半个小时候后,用水冲洗至中性,可改善保留时间不一致、峰形差等状况。
这一过程需要注意的是,活化后要水洗至中性,否则会引起聚合物峰成倍增大。
实际操作中发现用纯水冲洗很难完全洗去填料中的氢氧化钠,可行的方法是适当水洗后用缓冲盐流动相继续冲洗,可完全消除氢氧化钠残留的影响。