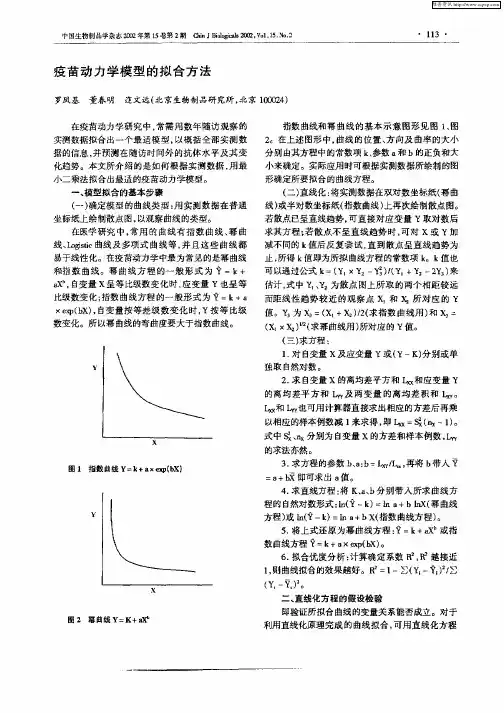
动力学方程拟合模型.
- 格式:doc
- 大小:506.45 KB
- 文档页数:8

动力学方程拟合模型动力学方程拟合模型主要分为幂函数型模型和双曲线型模型。
在幂函数型动力学方程中,温度和浓度被认为是独立地影响反应速率的,可以表示为:在双曲线型动力方程中强调模型方程中的吸附常数不能靠单独测定吸附性质来确定,而必须和反应速率常数一起由反应动力学实验确定。
这说明模型方程中的吸附平衡常数并不是真正的吸附平衡常数,模型假设的反应机理和实际反应机理也会有相当的距离。
双曲线型动力学方程的一般表达形式为上述两类动力学模型都具有很强的拟合实验数据的能力,都既可用于均相反应体系,也可用于非均相反应体系。
对气固相催化反应过程,幂函数型动力学方程可由捷姆金的非均匀表面吸附理论导出,但更常见的是将它作为一种纯经验的关联方式去拟合反应动力学的实验数据。
虽然,在这种情况中幂函数型动力学方程不能提供关于反应机理的任何信息,但因为这种方程形式简单、参数数目少,通常也能足够精确地拟合实验数据,所以在非均相反应过程开发和工业反应器设计中还是得到了广泛的应用。
1.幂函数拟合刘晓青[1]等人研究了HNO3介质中TiAP萃取Th(Ⅳ)的动力学模式和萃取动力学反应速率方程。
对于本萃取体系,由反应速率方程的一般形式可知:可用孤立变量法求得各反应物的分反应级数a、b与c,从而确立萃取动力学方程。
第一步:分级数的求算1.求a固定反应物中TiAP和HNO3的浓度,当TiAP的浓度远远大于体系中Th的初始浓度时,可以认为体系中TiAP浓度在整个萃取过程中没有变化而为一定値,则速率方程可以简化为两边取对数后得:ln{-d[Th-]/dt}=aln[Th]+ln1,用ln{-d[Th-]/dt}对ln[Th]作图得到一条直线(r=0.9973),其斜率即为a。
结果如图1所示,从图中可知斜率为1.05,即此动力学速率方程中Th(Ⅳ)的分反应级数a=1.05。
2.求b和c同求Th(Ⅳ)分反应级数类似,固定反应物中Th(Ⅳ)和HNO3的浓度,则速率方程可以简化为固定反应物中Th(Ⅳ)和TiAP的浓度,则速率方程可以简化为画图可得:TiAP的分反应级数为1.77,HNO3的分反应级数为0.38。

化学反应器的数学模型及其控制序言化学反应器是化学工业生产的核心设备,其鲁棒性和可控性是影响生产质量和效益的重要因素。
本文将介绍化学反应器的数学模型及其控制策略,旨在为化学工业生产和控制系统的优化提供参考。
一、化学反应器的数学模型化学反应器的数学模型是基于质量守恒、能量守恒和化学反应动力学等理论进行建立的。
其中,最常用的模型是连续拟合模型和分布参数模型。
1. 连续拟合模型连续拟合模型采用宏观平衡方程和动力学方程对反应器系统进行描述。
宏观平衡方程包括质量平衡和能量平衡两部分。
动力学方程则描述了物料在反应过程中的转化速率。
该模型通常采用微分方程组进行求解。
以催化剂颗粒床反应器为例,其数学模型如下:(1)质量平衡方程:$$\frac{\partial(\rho C W)}{\partial t}+\frac{\partial(\rho C W u)}{\partial x}=0$$(2)能量平衡方程:$$\frac{\partial(\rho C_p W T)}{\partial t}+\frac{\partial(\rho C_p W T u)}{\partial x}=\frac{\partial}{\partial x}(\lambda\frac{\partialT}{\partial x})+r\Delta H_R$$(3)物料转化速率方程:$$r=k(C_{A,f}-C_A)^n$$其中,$\rho$ 为颗粒床密度,$C$ 为反应物质浓度,$W$ 为颗粒床体积,$u$ 为颗粒床内流速,$x$ 为颗粒床内径向坐标,$T$ 为颗粒床内温度,$C_p$ 为热容,$\lambda$ 为导热系数,$r$ 为反应速率,$k$ 为反应速率常数,$n$ 为阶数,$\DeltaH_R$ 为反应焓变,$C_{A,f}$ 为反应物质浓度。
2. 分布参数模型分布参数模型则是采用微小体积元方法对反应器系统进行离散化,将反应器分为若干个微小体积,分别进行建模。

化学反应过程的计量学分析和反应动力学参数拟合方法化学反应过程是化学学科的核心领域之一,对于了解反应的本质和改进反应工艺具有重要意义。
在研究化学反应过程中,计量学分析和反应动力学参数拟合方法是两个关键概念。
计量学分析是指通过实验测定反应物和生成物的量,推导出化学方程式的反应系数。
反应动力学参数拟合方法则旨在研究反应速率与反应条件之间的关系。
计量学分析在化学反应过程中具有重要意义。
通过仔细测量反应物和生成物的物质量,可以确定化学方程式中的反应系数。
例如,对于涉及氧化还原反应的反应物A和B生成产物C和D的反应:aA + bB → cC + dD通过实验,我们可以测量反应物A和B的摩尔量,以及产物C和D的摩尔量。
根据质量守恒定律和摩尔量比例,我们可以计算出反应系数a、b、c和d的数值。
计量学分析提供了确定化学反应过程中物质转化的定量信息,为研究反应机制和改进反应工艺提供了基础。
而反应动力学参数拟合方法则关注反应速率与反应条件之间的关系。
反应速率是指单位时间内发生反应的物质转化率。
反应速率通常与反应物浓度、温度和催化剂等因素有关。
在研究反应动力学时,常用的方法是测量在不同反应条件下的反应速率,并将实验数据拟合到适当的数学模型中。
最常用的反应动力学模型是速率方程模型。
速率方程关联反应速率与反应物浓度的关系,其形式可由实验数据拟合得出。
例如,对于一级反应,其速率方程可以表示为:r = k[A]其中,r是反应速率,k是反应速率常数,[A]是反应物A的浓度。
通过实验测量不同浓度下的反应速率,可以求解出反应速率常数k的数值。
除了速率方程模型外,还有一些更复杂的模型可用于描述特殊类型的化学反应。
例如,对于涉及催化剂的反应,常用的模型是麦尔金-韦特模型。
该模型考虑反应物在催化剂表面上的吸附和解离过程,能更准确地描述反应动力学过程。
拟合实验数据到适当的数学模型中是确定反应动力学参数的关键一步。
常用的拟合方法包括最小二乘法和非线性回归分析。

matlab拟合动力学方程
MATLAB可以用于拟合动力学方程。
在MATLAB中,我们可以使用curve fitting工具箱来实现这个目标。
首先,我们需要收集我们的数据,并确定我们要拟合的动力学方程的类型。
例如,我们可以选择一阶动力学方程:dy/dt = -k*y,其中y是我们的输出变量,t是时间,k是动力学常数。
然后,我们可以使用MATLAB的curve fitting工具箱来拟合这个方程。
以下是一些步骤:
1. 导入数据:将我们收集的数据导入MATLAB环境。
确保数据已经存储为一个列向量,例如y和t。
2. 建立起始参数:根据我们的动力学方程,我们需要为k提供一个初始猜测值。
这个值可以根据我们的应用和经验来确定。
3. 建立模型:使用fittype函数创建一个模型对象,该对象表示我们要拟合的动力学方程。
4. 进行拟合:使用fit函数拟合我们的数据。
该函数将数据和模型作为参数,并返回包含拟合结果的对象。
5. 分析结果:我们可以通过访问拟合对象的属性来分析拟合结果,例如拟合参数的值和置信区间。
6. 可视化结果:使用plot函数绘制原始数据和拟合结果的图像,以便我们可以直观地评估拟合的质量。
通过这些步骤,我们可以使用MATLAB拟合动力学方程,并从拟合结果中获得我们感兴趣的参数值。

准一级和准二级动力学模型拟合参数
准一级动力学模型是指反应速率与反应物浓度的一次方成正比,其微分方程可以表示为:dy/dt=k[A],其中y为反应物的浓度,t为反应时间,k为反应速率常数,[A]为反应物的浓度。
该模型适用于反应物浓度较低、反应机理为单分子解离或反应中心为稀有物质的情况。
准二级动力学模型是指反应速率与反应物浓度的二次方成正比,其微分方程可以表示为:dy/dt=k[A]^2,其中y为反应物的浓度,t为反应时间,k为反应速率常数,[A]为反应物的浓度。
该模型适用于反应物浓度较高、反应机理为双分子解离或反应中心为富集物质的情况。
关于准一级和准二级动力学模型拟合参数,涉及到复杂的数学模型和计算,建议您请教专业人士。

一级动力学拟合
一级动力学拟合是探究某一系统行为方式的一种方式,它可以被用于解释这种行为方式的根本原因。
下面我们来一步步了解一级动力学拟合的具体内容和应用场景。
一、什么是一级动力学拟合?
一级动力学拟合是通过对某个系统所受到的外部刺激进行实验,并记录下系统响应的变化情况,然后将所得到的数据用一级动力学模型进行拟合并进行分析,以确定实验结果的相关特性与其所受到的刺激之间的关系。
二、一级动力学拟合的基本原理
一级动力学拟合是建立在对科学实验的基本原理上的,其具体的原理包括对输入与输出变量之间的关系进行建模,使用线性方程或非线性方程来拟合所得实验数据,以及选择适当的参数来解释实验结果。
三、一级动力学拟合的应用场景
1.经济学:一级动力学拟合可以被用于研究市场和货币政策对于经济系统的影响。
2.环境学:一级动力学拟合可以被用于预测和控制环境变化的影响,尤其是在探究气候变化的场景下。
3.生物学:一级动力学拟合可以帮助研究生物体的组织生长和代谢过
程以及受到外部环境变化的影响,用于预测疾病的发生和治疗效果。
四、一级动力学拟合的注意事项
1.对数据质量较低的实验, 一级动力学拟合可能会得到错误的解释。
2.适当的模型选择可以更好地解释实验数据和研究结果。
3.一级动力学拟合需要注意实验数据的采集和实验条件的准确性。
五、汇总
一级动力学拟合是一种探究系统行为方式的方法,其原理是通过对输入输出变量之间的关系建立线性方程或非线性方程,在实验中查看所得数据并进行适当的参数选择,从而解释实验结果。
它在经济学、环境学和生物学等领域有广泛的应用,但需要注意数据质量、模型选择和实验条件的准确性等问题。

origin拟合准二级动力学方程
Origin是一款科学绘图软件,可以用于数据分析和拟合。
准二级动力学方程是描述快速反应动力学的方程之一,其形式为:
d[A]/dt = k([A]0 - [A])^2
其中,[A]是反应物的浓度,t是时间,k是反应速率常数,[A]0是反应物的初始浓度。
在Origin中,拟合准二级动力学方程可以通过以下步骤完成:
1. 打开Origin软件,并导入数据文件。
2. 选择数据,并打开拟合窗口。
3. 在拟合窗口中选择“非线性拟合”选项。
4. 在拟合函数列表中选择准二级动力学方程。
5. 进行参数设置,包括起始参数、参数界限等。
6. 进行拟合,并查看拟合结果和拟合曲线。
7. 分析拟合结果,包括参数值、相关系数、拟合优度等。
需要注意的是,拟合准二级动力学方程需要有足够的数据点和反应速率常数的估计值,否则拟合结果可能不准确。
同时,拟合结果也需要进行统计分析和验证,
以确保其可靠性和可重复性。



化学反应动力学模型的参数估计化学反应动力学模型是描述化学反应速率的方程式,而模型参数的估计是建立反应动力学模型的关键步骤。
准确地估计反应动力学参数可以提高反应的预测能力和优化反应条件。
一、反应动力学模型介绍简单来说,化学反应速率的变化与反应中物质浓度之间的关系是反应动力学的研究范畴。
对于一般情况下,反应动力学模型可以被表示为:Rate = f(c1, c2, …, cn)这里,Rate是反应速率,ci是反应物或者中间体的浓度。
常见的反应动力学模型包括三种类型:零阶、一阶和二阶。
其中,零阶动力学指反应速率是不依赖反应物浓度的;一阶动力学指反应速率与某个反应物的浓度成正比;二阶动力学指反应速率与两个反应物的浓度成正比。
常见的反应动力学方程如下:零阶反应速率方程:Rate = k一阶反应速率方程:Rate = k[c]二阶反应速率方程:Rate = k[c1][c2]在这些方程中,k是速率常数,反映了反应速率与反应物浓度之间的比率关系,用于描述反应物浓度对于反应速率的影响。
二、反应动力学参数估计估计反应动力学模型的参数是反应动力学的研究关键之一,因为准确地估计参数可以提高模型预测的可信度和精度,有利于优化反应过程。
通常的,反应动力学模型中的速率常数k可以通过实验数据来估计。
在很多实验中,人们主要检测反应物的浓度随时间的变化,然后可以根据反应动力学模型方程来计算反应速率。
根据数据拟合反应动力学模型方程,可以得到速率常数k的估计值。
这个过程通常基于最小二乘法的理论框架来实现,其中模型拟合的目标是最小化测量误差的平方和。
在拟合反应动力学模型时,有几个要注意的问题:•确定反应物的浓度区域: 反应物浓度的选择可能会影响拟合反应动力学参数的精度。
• 选择测量模式:选择最合适的测量模式有利于提高数据质量。
• 考虑实验误差:在拟合反应动力学模型时,需要考虑测量误差的影响,以提高反应动力学数据的可靠性。
三、应用反应动力学模型的例子许多反应动力学模型已经应用于化学反应、生物反应、环境反应和工程反应等领域。
酶学反应动力学模型的模拟与参数拟合酶学反应动力学模型是描述生物化学反应速率和反应物转化的一个重要工具。
这种模型可以用来研究生化反应的动力学过程,揭示反应速率与温度、pH值、浓度等因素之间的关系。
基于酶学反应动力学模型的模拟与参数拟合可以深入理解生化反应的机理,并为实验设计和数据分析提供指导。
本文将介绍酶学反应动力学模型的基本原理、模拟方法和参数拟合策略。
一、酶学反应动力学模型的基本原理酶学反应动力学模型是描述酶促反应(即在酶催化下进行的生化反应)的速率随反应物浓度(也称底物浓度)的变化关系。
这种模型的基础是酶的底物饱和性(即酶对底物的亲和力的极限大小),以及酶反应速率随底物浓度变化的非线性关系。
常用的酶学反应动力学模型包括米氏方程、互补方程和希尔方程等。
米氏方程是最常用的酶学模型,用公式表示为:V = Vmax [S] / (Km + [S])其中,V是反应速率(即底物消耗或产物生成的速度),Vmax是酶最大反应速率(当底物浓度无限大时反应达到极限速率),[S]是底物浓度,Km是半饱和常数(即[底物] = Km 时速率达到Vmax的一半)。
米氏方程可以解释酶反应速率的非线性行为,因为随着底物浓度的增加,酶饱和度增加,进一步抑制了反应速率的增长。
二、酶学反应动力学模型的模拟方法酶学反应动力学模型的模拟方法包括分析解和数值解。
分析解是通过数学方法推导出理论表达式,求解析解可以获得模型的各种参数和关系。
数值解则是通过计算机模拟的方式求解微分方程,即在不同的底物浓度下计算出反应速率和产物浓度的大小和变化规律。
数值解方法适合于求解非线性微分方程或难以得到解析解的情况。
常用的数值解方法包括欧拉方法、龙格-库塔方法和辛普森法等。
这些方法都需要将微分方程转化为差分方程或积分形式来进行计算。
以欧拉方法为例,它的基本思路是利用微分方程将变化率转化为差分形式,通过一步一步的迭代来计算函数的值。
三、酶学反应动力学模型的参数拟合策略酶学反应动力学模型的参数拟合是将实验数据拟合到酶学模型中,以获得最优的模型参数。
生物化学反应动力学方程的计算方法生物化学反应动力学方程是生命科学中应用广泛的一种定量描述化学反应速率的方程式,它可以揭示化学反应的动力学规律,反应速率与反应物浓度的关系等重要信息。
本文将探讨生物化学反应动力学方程的计算方法,帮助读者深入了解这一领域的知识。
1. 动力学方程基础反应速率与浓度的关系是生物化学反应动力学方程的核心内容,此关系可总结为速率公式:R = k[A]^m[B]^n其中, R是反应速率;k是速率常数,表示单位时间内每个反应物分子参与反应的次数;[A]和[B]是反应物的浓度;m和n是反应物的反应次数,也叫反应级数。
反应级数指的是反应中各反应物参与反应的次数,一般用大写字母表示,比如A + B --> C的反应级数为2。
此外,反应级数还可说明反应机理和反应某一阶段速率的限制因素,如下所示:m = n = 0:零级反应,速率与反应物浓度无关。
m = 1,n = 0:一级反应,仅一个反应物参与反应,速率与其浓度成正比关系。
m = 2,n = 0:二级反应,两个反应物参与反应,速率与两者浓度的平方成正比关系。
m = 1,n = 1:二级反应,两个反应物参与反应,速率与两者浓度的积成正比关系。
2. 伦逻兹定理伦逻兹定理是描述分子碰撞频率与反应速率关系的理论,可以被应用于生物化学反应动力学研究中。
这一定理的核心是碰撞次数,可以表示为:Z = 4πN[(m1m2)/MKT]^(1/2)d^2v其中,N是分子密度;m1和m2是反应物分子的质量;K是玻尔兹曼常数;T是反应物的温度;d是反应物分子的直径;v是反应物分子的平均速率。
伦逻兹定理指出,分子碰撞频率与温度、浓度、反应物分子大小和形状等因素有关,这些因素均可影响化学反应速率。
在实际研究中,可以利用伦逻兹定理推导出反应物浓度与反应速率的关系式:R = k'Z[A][B]其中,k'是包含温度、分子大小和形状等修正因子的速率常数。
化学动力学模型化学动力学是研究化学反应速率和反应机理的学科。
通过建立化学动力学模型,可以对反应速率进行预测和分析,解释反应机理,并帮助设计和优化化学反应过程。
本文将介绍化学动力学模型的基本原理和常用方法。
一、化学反应速率方程化学反应速率是化学反应在单位时间内所消耗的物质量或生成的物质量与反应物浓度之间的关系。
用数学表达式表示为:v = k[A]^m[B]^n其中,v表示反应速率,k为速率常数,[A]和[B]分别表示反应物A和B的浓度,m和n为反应速率与浓度的指数,称为反应级数。
二、动力学模型的建立建立化学动力学模型的基本步骤如下:1. 确定反应机理:通过实验数据和理论分析推断出反应的基本步骤和中间产物。
2. 假设速率方程:根据反应机理假设反应速率方程,通常假设为速率限制步骤的反应。
根据反应级数和反应物浓度确定速率方程中的指数。
3. 决定速率常数:通过实验测定不同条件下的反应速率,使用非线性最小二乘法拟合实验数据,获得速率常数的数值。
4. 验证模型:将模型的预测结果与实验数据进行比较,验证模型的准确性和可靠性。
三、常用的1. 一阶反应模型:一阶反应指速率与反应物浓度的一次方成正比。
速率方程为:v = k[A]2. 二阶反应模型:二阶反应指速率与反应物浓度的二次方成正比。
速率方程为:v = k[A]^23. 伪一阶反应模型:当反应物A的浓度远远大于反应物B的浓度时,可以近似看作一阶反应。
速率方程为:v = k[A]4. 伪二阶反应模型:当反应物B的浓度远远大于反应物A的浓度时,可以近似看作二阶反应。
速率方程为:v = k[B]^25. 多分子反应模型:当反应物A和B同时参与反应,速率方程为:v = k[A]^m[B]^n四、应用案例化学动力学模型广泛应用于各个领域,例如:1. 化学工程:用于设计和优化化学反应过程的反应器尺寸和操作条件。
2. 药物研发:通过研究药物分解的速率,预测药物的稳定性和保存期限。
matlab拟合动力学参数
要在 MATLAB 中拟合动力学参数,可以使用 MATLAB 中的曲线拟合工具箱(Curve Fitting Toolbox)。
下面是一种拟合动力学参数的基本流程:
1. 确定动力学模型的结构和参数。
例如,如果要拟合一个一阶动力学模型,可以使用以下形式的方程:y = a*exp(b*x) + c,其中a、b、c是待确定的参数。
2. 准备数据。
将实验或观测数据以向量的形式导入到MATLAB 中。
假设数据的自变量为x,因变量为y。
3. 使用曲线拟合工具箱提供的拟合函数进行参数拟合。
在MATLAB 命令窗口中输入“cftool”打开曲线拟合工具箱界面。
选择“从文件导入数据”或手动输入数据。
在工具箱界面中,选择适当的拟合类型(例如指数型)和模型(例如一阶动力学模型),然后点击“拟合”按钮。
4. 分析拟合结果。
在拟合完成后,曲线拟合工具箱将显示拟合曲线和拟合参数。
可以分析拟合结果,比如查看拟合曲线与原始数据的拟合程度,以及拟合参数的值和相关统计信息。
5. 获取拟合参数。
在拟合结果中,可以找到拟合参数的值。
将这些值用于进一步的分析或应用。
需要注意的是,正确选择动力学模型的结构和参数是关键。
如
果对动力学模型有疑问或不确定,可以查阅相关文献或咨询专业领域的专家。
化学反应速率的动力学模型的理论解释化学反应速率是描述化学反应过程中物质浓度变化率的物理量。
在化学反应动力学中,为了解释反应速率与反应物浓度的关系,科学家提出了多种动力学模型。
本文将对几种常见的动力学模型进行理论解释。
一、零级动力学模型零级动力学模型适用于指数上升或下降的反应速率情况。
该模型假设反应速率与反应物浓度无关,即反应速率为恒定值。
这意味着反应物浓度的变化不会影响反应速率,而是由其他因素所决定。
零级反应速率方程可以表示为R = k。
二、一级动力学模型一级动力学模型适用于反应速率与反应物浓度成正比的情况。
该模型假设反应速率与反应物浓度之间存在线性关系,即反应速率与反应物浓度呈一次函数关系。
一级反应速率方程可以表示为R = k[A],其中R为反应速率,k为速率常数,[A]为反应物A的浓度。
三、二级动力学模型二级动力学模型适用于反应速率与反应物浓度成平方关系的情况。
该模型假设反应速率与反应物浓度之间存在二次函数关系。
二级反应速率方程可以表示为R = k[A]^2,其中R为反应速率,k为速率常数,[A]为反应物A的浓度。
四、复合反应动力学模型复合反应动力学模型适用于复杂的反应速率与反应物浓度关系。
该模型可以由多种动力学模型的组合来表示。
例如,一个反应可以同时遵循一级和二级反应动力学。
复合反应动力学模型的具体形式将取决于反应的特殊情况和实验数据。
动力学模型的选择取决于具体的化学反应特征和研究目的。
科学家通过实验数据的分析和模型拟合来确定最适合描述反应速率的动力学模型。
其中,速率常数k是一个重要参数,表示了反应的速率和反应物浓度之间的关系。
除了上述介绍的几种常见动力学模型外,还存在许多其他模型用于解释不同类型的化学反应速率。
这些模型基于不同的假设和数学关系,可以更好地描述特定的化学反应动力学。
根据实际研究需求,科学家可以选择合适的模型来解释化学反应速率的变化规律。
总结起来,化学反应速率的动力学模型提供了一种理论解释反应速率与反应物浓度之间关系的方法。
动力学方程拟合模型
动力学方程拟合模型主要分为幂函数型模型和双曲线型模型。
在幂函数型动力学方程中,温度和浓度被认为是独立地影响反应速率的,可以表示为:
在双曲线型动力方程中强调模型方程中的吸附常数不能靠单独测定吸附性质来确定,而必须和反应速率常数一起由反应动力学实验确定。
这说明模型方程中的吸附平衡常数并不是真正的吸附平衡常数,模型假设的反应机理和实际反应机理也会有相当的距离。
双曲线型动力学方程的一般表达形式为
上述两类动力学模型都具有很强的拟合实验数据的能力,都既可用于均相反应体系,也可用于非均相反应体系。
对气固相催化反应过程,幂函数型动力学方程可由捷姆金的非均匀表面吸附理论导出,但更常见的是将它作为一种纯经验的关联方式去拟合反应动力学的实验数据。
虽然,在这种情况中幂函数型动力学方程不能提供关于反应机理的任何信息,但因为这种方程形式简单、参数数目少,通常也能足够精确地拟合实验数据,所以在非均相反应过程开发和工业反应器设计中还是得到了广泛的应用。
1.幂函数拟合
刘晓青[1]等人研究了HNO3介质中TiAP萃取Th(Ⅳ)的动力学模式和萃取动力学反应速率方程。
对于本萃取体系,由反应速率方程的一般形式可知:
可用孤立变量法求得各反应物的分反应级数a、b与c,从而确立萃取动力学方程。
第一步:分级数的求算
1.求a
固定反应物中TiAP和HNO3的浓度,
当TiAP的浓度远远大于体系中Th的初始浓
度时,可以认为体系中TiAP浓度在整个萃
取过程中没有变化而为一定値,则速率方程
可以简化为
两边取对数后得:
ln{-d[Th-]/dt}=aln[Th]+ln1,用ln{-d[Th-]/dt}
对ln[Th]作图得到一条直线(r=0.9973),其斜率即为a。
结果如图1所示,从图中可知斜率为1.05,即此动力学速率方程中Th(Ⅳ)的分反应级数a=1.05。
2.求b和c
同求Th(Ⅳ)分反应级数类似,固定反应物中Th(Ⅳ)和HNO3的浓度,则速率方程可以简化为
固定反应物中Th(Ⅳ)和TiAP的浓度,则速率方程可以简化为
画图可得:
TiAP的分反应级数为1.77,HNO3的分反应级数为0.38。
第二步:写出反应反应速率方程
则293K时,该反应的平均速率常数k为1.6*10-2(mol/L)-2.2·s-1。
2.双曲线拟合
双曲线型反应动力学方程是由Hinshelwood在研究气固相催化反应动力学时,根据Langmuir的均匀表面吸附理论导出的,其后Hougen和Watson用此模型成功地处理了许多气固相催化反应,使它成为一种广泛应用的方法。
因此,双曲线型动力学方程又被称为Langmuir-Hin-shelwood方程或Hougen-Watson方程。
王志良[2]等人用半连续式无梯度反应器在130~210 ℃范围内研究了苯与乙烯在FX-02沸石催化剂上烷基化反应的本征动力学。
应用改进的Gauss-Newton 法对常微分形式的动力学模型进行了参数估值,得到了双曲函数形式的本征动力学方程。
体系的反应情况如下:
乙烯( E)、苯( B)、乙苯( EB)、二乙苯( DEB)
根据Langmuir-Hinshelwood 机理, 对上述两个反应用如下的动力学模型来表示:
序贯试验设计选用最小体积判别式( MVD ):
对ks1、ks- 1、ks2、ks- 2、KE、KB、KEB、KDEB八个参数采用改进的Gauss-Newton 法,直接对常微分形式的动力学模型进行参数估值,目标函数由最小二乘估值准则确定:
经过动力学的预试验和序贯试验后,下图给出了参数估值的相对置信区间大小与试验次数的关系。
可以看出,当进行5次序贯试验后,Δ-1/2的值迅速下降, 表
明试验点的安排较合理,在有效的试验次数内使参数的估值达到了相当高的置信度。
参数估计是在序贯试验下进行的,对参数的联合置信区间检验结果证明,参数具有很高的精度。
模型检验其方差分析结果表明,计算值与试验数据的相符性良好, 残差分析进一步证明模型无缺陷。
3.其他算法在动力学方程中的应用
在查找文献的时候,找到多篇与动力学有关的文献采用了不同的算法对动力学参数进行拟合估算。
3.1遗传算法
遗传算法(Genetic Algorithm)是模拟达尔文生物进化论的自然选择和遗传学机理的生物进化过程的计算模型,是一种通过模拟自然进化过程搜索最优解的方法。
黄晓峰[3]等用改进的实数编码遗传算法进行了估计反应动力学参数的研究,提出了一种优化分布线性交叉操作策略,使子代个体在搜索空间内达到均匀分布,从而提高了搜索的效率。
作者用这种改进的实数编码遗传算法进行了正丁烷选择氧化反应动力学参数的估计。
正丁烷在VPO催化剂上选择氧化制顺酐,是催化晶格氧参与催化循环、按照氧化还原( RE-DOX) 机理进行的重要烃类选择氧化反应,其简化后的基元反应序列为:
R 和X 分别代表还原态和氧化态催化剂,B 为正丁烷,MA 为顺酐。
主要操作策略和控制参数为:适应度线性调整、带最优个体保存的期望值选
择, 优化分布的线性交叉操作和连续变异操作。
种群数目N =50 , 交叉概率Pc =0.8 , 变异概率Pm =0.05 , 交叉系数α=2.0 。
优化问题描述为估计反应速率常数K 1 、K 2 或活化能E 1 、E 2 与指前因子k01 、k 02 , 以使反应速率的估计值rBE 与测量值rBM 的偏差平方和RSS 极小化:
最后结果如下,表现出较高的精度。
3.2蒙特卡罗法
詹晓力[4]等利用蒙特卡罗方法模拟计算了化学反应动力学参数,由基元反应确定蒙特卡罗模拟的具体做法,将蒙特卡罗方法的模拟结果与动力学实验结果进行比较,根据比较结果自动调整和优化动力学参数,从而无需事先确定动力学方程,即可有效地估算各种化学反应的动力学参数。
用该方法模拟Mo-Bi 系丙烯氨氧化催化剂上的氨分解基元反应。
无丙烯存在下的氨分解基元反应如下:
下图给出了用蒙特卡罗方法对该问题进行模拟的结果,m 为催化剂质量,nA0为初始氨物质的量,c R为转化率,r 为速率。
从图中可见,按估算的动力学参数所计算的转化率-时间和反应速率-时间曲线与实验数据吻合得较好。
总结
化学反应的机理通常是十分复杂的。
一些看起来相当简单的反应的机理至今也没有完全搞清。
因此,不论是双曲线型模型还是幂函数型模型,都只是可以用来拟合反应动力学实验数据的一种函数形式。
由于这两种方程在数学上的适应性极强,对同一组实验数据可同时用这两种方程拟合的例子也是屡见不鲜的。
从这个意义上讲,目前工程上应用的绝大多数动力学模型都不是机理模型,在原实验范围之外作大幅度的外推都是有风险的。
参考文献
[1]刘晓青等.HNO3介质中TiAP萃取Th(Ⅳ)的动力学研究[J].四川大学学报,2014,51(6):1249-1254.
[2]王志良等.FX-02沸石催化剂上苯与乙烯烷基化的反应动力学[J].石油炼制与化工,1999,30(2):52-55.
[3]黄晓峰,潘立登,陈标华,等.用改进的实数编码遗传算法估计反应动力学参数[J].高校化学工程学报,1999,13( 1) : 50-55.
[4]詹晓力,罗正鸿,陈丰秋,等.基于Monte Carlo 模拟的化学反应动力学参数估算[J].高
等学校化学学报,2003,24( 8) : 1511-1514.。