分子构建转化鉴定(分子生物学实验)
- 格式:pdf
- 大小:227.97 KB
- 文档页数:6

生物化学及分子生物学实验考试-图文一、实验原理步骤:1、分子克隆总流程:分子克隆:在分子水平上提供一种纯化和扩增特定DNA片段的方法。
常含有目的基因,用体外重组方法将它们插入克隆载体,形成重组克隆载体,通过转化与转导的方式,引入适合的寄主体内得到复制与扩增,然后再从筛选的寄主细胞内分离提纯所需的克隆载体,可以得到插入DNA的许多拷贝,从而获得目的基因的扩增。
流程:(1)带有目的基因的DNA片段的获得——PCR引物设计,PCR获得。
(2)重组DNA分子的构建——与载体酶切连接。
(3)重组DNA分子的转化和重组克隆的筛选——转化,培养,筛选(4)检测——菌落PCR检测,粗提质粒检测,酶切检测,精提质粒测序。
2、质粒DNA的提取及鉴定一一碱裂解法原理:根据共价闭合环状质粒DNA和线性染色体DNA在拓扑学上的差异来分离质粒DNA。
在pH12.0-12.5,线性的DNA双螺旋结构解开而被变性,尽管在这样的条件下,质粒DNA的氢键断裂,但两条互补链仍紧密结合。
当加入pH4.8的KAc高盐缓冲液中和pH至中性时,共价闭合环状的质粒DNA复性迅速而准确,而线性的染色体DNA的两条互补链彼此已完全分开,复性慢,缠绕形成网状结构。
通过离心,染色体DNA与RNA、蛋白质-SDS复合物等一起絮状沉淀下来而被除去。
步骤:1、细菌培养和收集①将3ml含Kan的LB液体培养基加入到试管中,接入含质粒的大肠杆菌,37°C振荡培养过夜。
(已完成)②取2.5mL培养物倒入微量离心管(EP管)中,10000rpm离心lmin,吸去培养液。
2、细菌裂解及质粒的提取③将细胞沉淀悬浮于100ul溶液I中,充分震荡混匀。
④加200ul溶液II(新鲜配制),盖紧管盖,轻轻混匀内容物,将离心管放冰上5min。
⑤加150ul溶液III(冰上预冷),盖紧管口,轻轻颠倒数次使混匀。
冰上放置10min。
⑥12000rpm离心10min,将上清转至另一EP管(作好标记)中。

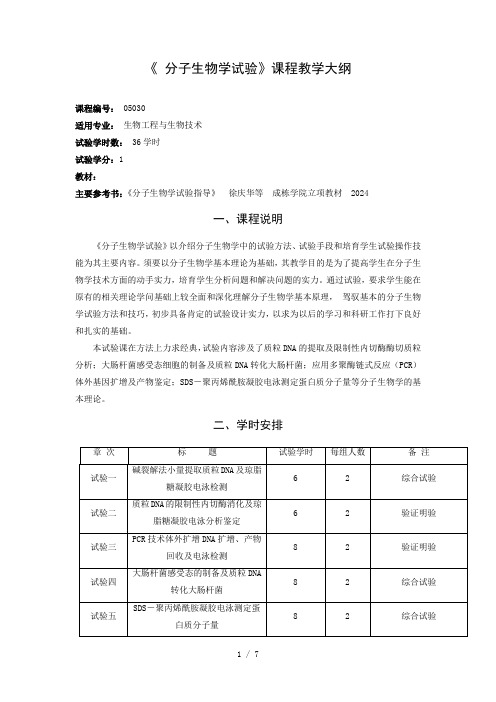
《分子生物学试验》课程教学大纲课程编号: 05030适用专业:生物工程与生物技术试验学时数:36学时试验学分:1教材:主要参考书:《分子生物学试验指导》徐庆华等成栋学院立项教材 2024一、课程说明《分子生物学试验》以介绍分子生物学中的试验方法、试验手段和培育学生试验操作技能为其主要内容。
须要以分子生物学基本理论为基础,其教学目的是为了提高学生在分子生物学技术方面的动手实力,培育学生分析问题和解决问题的实力。
通过试验,要求学生能在原有的相关理论学问基础上较全面和深化理解分子生物学基本原理,驾驭基本的分子生物学试验方法和技巧,初步具备肯定的试验设计实力,以求为以后的学习和科研工作打下良好和扎实的基础。
本试验课在方法上力求经典,试验内容涉及了质粒DNA的提取及限制性内切酶酶切质粒分析;大肠杆菌感受态细胞的制备及质粒DNA转化大肠杆菌;应用多聚酶链式反应(PCR)体外基因扩增及产物鉴定;SDS-聚丙烯酰胺凝胶电泳测定蛋白质分子量等分子生物学的基本理论。
二、学时安排三、教学内容及教学基本要求试验一碱裂解法小量提取质粒DNA及琼脂糖凝胶电泳检测一、试验特点试验类型:综合试验类别:专业基础安排学时:6 每组人数:2二、试验目的1、驾驭碱裂解法小量提取质粒DNA和试剂盒提取质粒DNA的原理和方法。
2、驾驭琼脂糖凝胶的制备及外源DNA的检测原理。
3、了解质粒DNA的粗略定量方法。
三、试验内容提要1、将单菌落接种于3m1含相应抗生素的LB培育基中,37℃摇菌过夜;2、12,000rpm离心30sec,收集菌体;3、加200u1溶液I(含RNaseA 100ug/ml ),振荡悬浮菌体;4、加200ul新配制的溶液II,颠倒混匀;5、溶液澄清后马上加入预冷的200u1溶液III混匀后冰上放置5~10 min;6、4℃、12,000rpm离心15min,取上清,加0.7倍异丙醇混匀,室温静置5min;7、12,000rpm离心10min,70%乙醇洗涤沉淀,抽干后溶于适量水或TE,-20℃保存备用。

分子生物学实验报告重组质粒的pcr验证(目的基因的pcr扩增)姓名:xxx学号:xxx专业:xxx界别:xxx同组者:xxxx一.实验目的(1)自学掌控pcr反应的基本原理和实验技术。
(2)了解引物设计的基本要求。
1.pcr基本原理聚合酶链式反应(polymerasechainreaction),简称pcr,是一种分子生物学技术,用于在体外快速扩增dna,类似dna的细胞内复制过程:由一对引物介导,通过温度的调节,使双链dna变性为单链dna、单链dna能与引物复性(退火)成为引物-dna单链复合物、以及在dntps存在下dna聚合酶能使引物沿单链模板延伸成为双链dna(引物的延伸);这种热变性-复性-延伸的过程,就是一个pcr循环;一般通过20-30个循环之后,就可获得大量的要扩增的dna片段。
pcr技术的基本原理类似dna的天然激活过程,其特异性依赖与靶序列两端优势互补的寡核苷酸引物。
pcr由变性--淬火--延展三个基本反应步骤形成:①模板dna的变性:模板dna经加热至90~95℃左右一定时间后,使模板dna双链或经pcr扩增形成的双链dna解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板dna与引物的淬火(复性):模板dna经冷却变性成单链后,温度降到55℃左右,引物与模板dna单链的优势互补序列接合融合;③引物的延伸:dna模板--引物结合物在taqdna聚合酶的作用下,以dntp为反应原料,靶序列为模板,按碱基互补配对与半保留复制原理,合成一条新的与模板dna链互补的复制链。
经过“变性—淬火—延展”三个过程就可以赢得代莱dna分子,而且这种崭新dna分子又可以沦为下次循环的模板。
因此,变性、淬火和延展循环反复25~30次后,即可从少量的模板dna中扩充出来大量的目的产物。
pcr引物设计的目的是为了找到一对合适的核苷酸片段,使其能有效地扩增模板dna序列,因此,引物的优劣直接关系到pcr的特异性与成功与否。

分子生物学实验分子生物学实验是研究生命分子结构和功能的基础。
分子生物学的研究对象是生命体内的DNA、RNA、蛋白质等生物分子,通过实验可深入了解生命分子的结构及其功能,为治疗疾病等产生重要的科研意义。
本文将着重介绍PCR扩增、基因克隆、蛋白质表达和酶联免疫吸附实验等分子生物学实验。
PCR扩增PCR扩增是分子生物学领域中使用最广泛的实验技术之一。
PCR扩增技术是通过不断的复制,使DNA分子增多。
PCR主要操作步骤包括:DNA样品提取、模板DNA扩增、DNA回收纯化和定量检测等。
PCR扩增实验分为两部分:第一部分是制备PCR反应混合物,包括模板DNA、引物(Primer)、反应缓冲液、酶、脱离剂、种子液等。
制备反应混合物的过程中,需要将所有的试剂按照一定比例混合后,将混合液均匀吸入PCR管内。
第二部分是PCR反应实验本身,包括控制PCR反应温度变化、确保每个PCR 反应的时间和温度一致、等等。
PCR反应时间通常在2-6小时之间,取决于所扩增的DNA片段的长度和针对不同目标所使用的引物。
基因克隆基因克隆是利用重组DNA技术将外源DNA和载体DNA组装重组成新的DNA分子,然后通过转化等方式将新的DNA分子转移到感受态宿主细胞中,使得新的DNA分子被细胞所接受并复制,从而达到克隆目的的一种实验方法。
基因克隆实验的主要操作步骤包括:选取适当的载体,将所需要的外源DNA和载体DNA连接起来组成重组DNA分子,将重组DNA分子转化至感受态宿主细胞中,最后从发酵液中提取所需的目标DNA分子。
基因克隆技术在分子生物学实验研究中起到了重要的作用,使得我们能够制备大量目标分子用于进一步的研究。
蛋白质表达蛋白质表达是一种将蛋白质合成出来的实验技术。
蛋白质表达技术的主要目的是利用外源基因的扩增技术,将目标蛋白表达并纯化出来,从而进一步深入了解蛋白质的结构及其功能。
蛋白质表达实验的主要操作步骤是:蛋白质的基因落库、基因克隆到重组基因载体中、启动子、子纯化表达及活性鉴定等。

分子生物学实验指导(总45页) -CAL-FENGHAI.-(YICAI)-Company One1-CAL-本页仅作为文档封面,使用请直接删除分子生物学实验指导分子生物学实验课程的内容是从亚克隆一个基因直到最终表达出该基因产物的一个完整的科研项目。
以表达纳豆激酶为例,从已经克隆在pMD-18-T 载体上的纳豆激酶酶原基因上用PCR扩增出837bp的纳豆激酶成熟肽基因片段,与T载体连接后转入DH5菌中筛选鉴定,然后用双酶切下纳豆激酶成熟肽基因片段,插入带有6个组氨酸标签的原核表达载体pET-Trx,在表达菌BL(21)DE3中诱导表达出纳豆激酶蛋白质并用SDS-PAGE鉴定。
整套实验围绕纳豆激酶基因的亚克隆、重组、转化、筛选直到表达这一研究顺序进行操作。
其间共涉及学习约11种基因操作的基本技术,我们按这些操作技术出现的先后顺序分别编为实验一至实验十一。
各个实验之间具有很强的逻辑性和连贯性,前一个实验的结果就是下一个实验的原料。
整套实验设计实际上是一个基因操作实验平台,随时能根据需要更改外源基因和表达载体或进一步扩充实验内容。
我们在每年的教学实践中会不断增加目的基因和表达载体的种类,以便于学生实验小组能够从事不同的项目,减少雷同,增加兴趣。
分子生物学实验的宗旨是让学生在独立实践操作中学习分子生物学的基本研究方法和体会分子生物学研究的严密逻辑和科研理念。
实验教学的组织方式是科研项目式管理,即在教师的总体指导下,在给定的实验材料和实验条件的基础上,让学生独立思考、自行规划实验流程、制定实验计划、准备实验材料、动手操作、整理和分析实验结果,最后完成实验项目,写出实验论文。
因此学生在实验前要认真预习实验内容,结合学过的分子生物学原理,弄懂每一步实验的目的和原理,了解实验的内容和总体实验方案,写出分子生物学大实验《开题报告》,交教师审阅和讨论修改后才能进入实验室操作。
在实验过程中,教师不干涉学生的实验安排和操作程序,仅以导师的身份对学生的实验操作进行现场巡回指导、回答学生的疑问、为学生示范一些高档实验仪器和软件的使用,提供公用的试剂(如酶)等,并对学生的每一步实验结果进行评价和把关。
实验:质粒DNA的制备、鉴定、转化及目标基因的诱导表达——参考《分子生物学实验》等一、实验目的1. 了解质粒DNA的性质,掌握碱裂解法提取质粒的方法;2. 了解电泳在生物大分子中分离和鉴定的基本原理,掌握琼脂糖凝胶的制备以及琼脂糖凝胶电泳在DNA片段分离中的应用方法;3. 了解感受态细胞的制备方法,学习DNA化学转化方法的过程;4. 掌握蛋白质的诱导表达方法及聚丙烯酰胺凝胶电泳在蛋白质鉴定中的应用。
二、实验原理(一)质粒DNA的提取-碱裂解法细菌质粒是一类双链、闭合环状的DNA,大小范围从1kb至200kb以上不等。
各种质粒都是存在于细胞质中、可以独立于细胞染色体之外的自主复制的遗传成份。
通常情况下它可持续稳定地处于染色体外的游离状态,但在一定条件下也会可逆地整合到寄主染色体上,随着染色体的复制而复制,并通过细胞分裂传递到后代。
一般分离质粒DNA的方法都包括3个步骤:1. 培养细菌,使质粒DNA大量扩增;2.收集和裂解细菌;3. 分离和纯化质粒DNA。
提取质粒DNA的方法很多,其中常用的方法有碱裂解法、煮沸法、SDS法、羟基磷灰石层析法等。
在实际操作中可以根据宿主菌株类型、质粒分子大小、碱基组成和结构等特点以及质粒DNA的用途进行选择。
本实验使用碱裂解法提取质粒DNA。
碱裂解法提取质粒DNA是根据共价闭合环状质粒DNA和线性染色体DNA在拓扑学上的差异来分离质粒DNA。
在pH值介于12.0-12.5这个狭窄的范围内,线性的DNA双螺旋结构解开而被变性,尽管在这样的条件下,共价闭环质粒DNA的氢键会被断裂,但两条互补链彼此相互盘绕,仍会紧密地结合在一起。
当加入pH4.8乙酸钾高盐缓冲液恢复pH至中性时,因为共价闭合环状的质粒DNA的两条互补链仍保持在一起,因此复性迅速而准确,而线性的染色体DNA的两条互补链彼此已完全分开,复性就不会那么迅速而准确,它们相互缠绕形成不溶性网状结构,而复性的质粒DNA恢复原来构型,保持可溶性状态。
分子生物学实验报告慕蓝有志班梦想学院目录实验一细菌的培养 (2)实验二质粒DNA的提取 (4)实验三琼脂糖凝胶电泳法检测DNA (7)实验四质粒DNA酶切及琼脂糖电泳分析鉴定 (9)实验五聚合酶链反应(PCR)技术体外扩增DNA (11)实验六植物基因组DNA提取、酶切及电泳分析 (14)实验七RNA分离与纯化 (17)实验八RT-PCR扩增目的基因cDNA (19)实验九质粒载体和外源DNA的连接反应 (21)实验十感受态细胞的制备及转化 (23)实验十一克隆的筛选和快速鉴定 (25)实验十二地高辛标记的Southern杂交 (27)实验十三阿拉伯糖诱导绿色荧光蛋白的表达 (31)思考题 (32)分子实验心得总结 (33)实验一细菌的培养一、目的学习细菌的培养方法及培养基的配置。
二、原理在基因工程实验和分子生物学实验中,细菌是不可缺少的实验材料。
质粒的保存、增殖和转化;基因文库的建立等都离不开细菌。
特别是常用的大肠杆菌。
大肠杆菌是含有长约3000kb的环状染色体的棒状细胞。
它能在仅含碳水化合物和提供氮、磷和微量元素的无机盐的培养基上快速生长。
当大肠杆菌在培养基中培养时,其开始裂殖前,先进入一个滞后期。
然后进入对数生长期,以20~30min复制一代的速度增殖。
最后,当培养基中的营养成分和氧耗尽或当培养基中废物的含量达到抑制细菌的快速生长的浓度时,菌体密度就达到一个比较恒定的值,这一时期叫做细菌生长的饱和期。
此时菌体密度可达到1×109~2×109/mL。
培养基可以是固体的培养基,也可以是液体培养基。
实验室中最常用的是LB培养基。
三、实验材料、试剂与主要仪器(一)实验材料大肠杆菌(二)试剂1. 胰蛋白胨2. 酵母提取物3. 氯化钠4. 1mol/L NaOH5. 琼脂粉6. 抗生素(氨苄青霉素、卡那霉素等)(三)仪器1. 培养皿2. 带帽试管3. 涂布器4. 灭菌锅5. 无菌操作台(含酒精灯、接种环、灭菌牙签等)6. 恒温摇床四、操作步骤(一)LB培养基的配制配制每升培养基,应在950m1去离子水中加入:细菌培养用胰蛋白胨10g细菌培养用酵母提取物5gNaCl 10g摇动容器直至溶质完全溶解,用1mol/L NaOH调节pH位至7.0。
分子生物学实验教材:参考书:《分子克隆实验指南》(第三版)黄培堂等译科学出版社实验一﹑大肠杆菌感受态细胞的制备及转化一.[目的要求]通过本实验,掌握大肠杆菌感受态细胞的制备及转化的方法和技术。
二.[实验原理]细菌处于容易吸收外源DNA的状态叫感受态。
转化是指质粒或以它为载体构建的重组子导入细菌的过程。
细菌处于0︒C ,CaCl2低渗溶液中,细胞膨胀成球形,外源DNA附着于细胞表面,经42︒C 短时间热击处理,促进细胞吸收DNA复合物。
将细胞放置在非选择性培养基中保温一段时间,促使在转化过程中获得的新表型如Amp+得到表达,然后将此细菌培养物涂在选择性培养基上进行培养,从而获得转化子。
三.[教学内容]1.感受态细胞的制备基础知识及方法简介2.CaCl2法制备DH5α或JM109感受态细胞,计算转化效率3.pUC19等质粒和重组产物转化大肠杆菌DH5α感受态细胞四.[实验材料和用品]E. coli DH5α菌株: Rˉ,Mˉ,Ampˉ;pBS质粒DNA,eppendorf管。
超净工作台、恒温摇床、生化培养箱、平皿、LB培养基、CaCl2 溶液、氨苄青霉素、等五.[实验步骤]一、受体菌的培养从LB平板上挑取新活化的E. coli DH5α单菌落,接种于3-5ml LB液体培养基中,37℃下振荡培养12小时左右,直至对数生长后期。
将该菌悬液以1:100-1:50的比例接种于100ml LB液体培养基中,37℃振荡培养2-3小时至OD600 =0.5左右。
二、感受态细胞的制备 ( CaCl2 法)1、将培养液转入离心管中,冰上放置10分钟,然后于4℃下3000g离心10分钟。
2、弃去上清,用预冷的0.05mol/L的CaCl2 溶液10ml轻轻悬浮细胞,冰上放置15-30分钟后,4℃下3000g离心10分钟。
3、弃去上清,加入4ml预冷含15%甘油的0.05mol/L的CaCl2 溶液,轻轻悬浮细胞,冰上放置几分钟,即成感受态细胞悬液。
DNA Molecule Recombination and Transformation Lin ChengyuBio 04 2010030007; Cooperator: Yuan Xiaowen1Introduction1.1Background InformationDNA Molecule Recombination refers to the process that the target DNA is put into thevector using biochemical methods. Then recombinant DNA is transferred into theorganism such as E.coli in order to get the product. Identification is needed to preventfalse positive clone.1.2Objectives(1)Learn the method of DNA purification by ethanol precipitation;(2)Learn the method of DNA subcloning;(3)Learn the CaCl2 method of E.coli competent cells preparation;(4)Learn the methods of transformation and clone selection;1.3Major Principles1.3.1Ethanol precipitationDNA molecule recombination needs purified DNA in order to get high successratio. In the experiment we use ethanol precipitation, because big DNAmolecule is insoluble in high ethanol concentration solution. And smallfragment of DNA can be eluted using this property, while the target DNA willbe in the pellet.1.3.2LigationConnect the 5’ end and 3’ end from two different DNA fragments using DNAligase. The process is shown in Figure 1.Figure 1 DNA ligation1.3.3 E.coli competent cells preparationNaturally, bacterial cells are not easy to allow foreign DNA to pass their cellwalls. However, when they are treated with some reagents, such as CaCl2, theircell walls will be altered which makes the foreign DNA enter the cells easily.Such cells are called "competent cells”.1.3.4TransformationThe process of introducing foreign DNA into the competent cells is called“transformation”. In this experiment, heat shock method is used to introduce theforeign DNA into the competent cells prepared by CaCl2 through heat shock.2Experiment Operation2.1MaterialPCR productpBluescript KS plasmidNEB 1kb DNA ladderBam HI, Hin dIIITable 1 Recognition Sequence and Cutting Site of Bam HI and Hin dIII1E.coli DH5α2.2Chemicals and Apparatus2.2.1SolutionsTable 2 SolutionsTable 2 Solutions (Continued)2.2.2Apparatus(1)Pipettes;(2)Eppendorf tubes;(3)Electrophoresis apparatus(4)UV gel imaging system(5)Water bath2.3Procedure2.3.1Restriction enzyme digestionAdd all the components into the Eppendorf tube following Table 3Table 3 Scheme of restriction enzyme digestionIncubate the tubes at 37 ℃for 1 h.2.3.2Ethanol precipitation(1)Add 2-fold (40 μL) absolute ethanol and 0.1-fold (2 μL) 3 M to theEppendorf tube;(2)Incubate at -20 ºC for 10 min;(3)Centrifuge at 12,000 rpm for 10 min, 4 ºC;(4)Discard the supernatant carefully, add 100 μL ethanol, and resuspend thepellet;(5)Centrifuge at 12,000 rpm for 10 min, 4 ºC;(6)Discard the supernatant thoroughly, and leave the tube uncovered for 5min;(7)Add 10 μL ddH2O to solute the target DNA;(8)Run agarose gel electrophoresis using 2 μL target DNA and 1 μL vector.2.3.3Ligation(1)Decide the concentration of target DNA and vector by comparing the twobands;(2)Add all components to the Eppendorf tube following Table 4;Table 4 Scheme of ligation2.3.4 E.coli competent cells preparation(1)Add 1.8 mL culture medium containing E.coli. to the Eppendorf tube;(2)Centrifuge at 4,000 rpm for 1 min, 4 ºC;(3)Discard the supernatant, and add another 1.8 mL culture mediumcontaining E.coli;(4)Centrifuge at 4,000 rpm for 10 min, 4 ºC;(5)Discard the supernatant, add 1 mL ice-cold 0.1 M CaCl2 and resuspend;(6)Centrifuge at 4,000 rpm for 10 min, 4 ºC;(7)Repeat Step 5 and 6;(8)Discard the supernatant, and 200 μL ice-cold 0.1 CaCl2 and resuspend;(9)Incubate on ice;2.3.5Transformation(1)Add 100 μL solution containing competent cells to an Eppendorf tube;(2)Add 10 μL recombinant plasmid;(3)Mix gently and incubate on ice for 20 min;(4)Heat shock at 42 ºC for 90 sec;(5)Put the tube on ice immediately, and incubate for 2 min;(6)Add 900 μL LB culture without antibiotic;(7)Incubate at 37 ºC for 20 min;(8)Incubate the cells on a new plate containing ampicillin, X-gal, and IPTG; 2.3.6Identification2.3.6.1Plasmid extraction and purification(1)Select a positive clone (white in this experiment) and put into the LBculture medium containing ampicillin;(2)Incubate at 37 ºC overnight;(3)Add 1.2 mL bacterial culture into an Eppendorf tube, and centrifuge at10,000 rpm for 1 min;(4)Discard the supernatant;(5)Repeat step 3 and 4;(6)Add 100 μL Solution I, and resuspend the pellet using pipette andvigorously vortexing;(7)Add 200 μL of freshly prepared Solution II, and mix the contents byinverting the tube rapidly five times;(8)Incubate on ice for 5 min;(9)Add 150 μL ice-cold solution III, and mix the contents by inverting the tubeseveral times;(10)I ncubate on ice for 3~5 min;(11)C entrifuge at 10,000 rpm for 7 min.(12)T ransfer the supernatant to a new Eppendorf tube, and add 1 mL absoluteethanol;(13)C entrifuge at 12,000 rpm for 5 min, and discard the supernatant;(14)A dd 500 μL 70 ethanol, and resuspend using pipette;(15)C entrifuge at 12,000 rpm for 5 min, and discard the supernatant;(16)I nvert the tube on a paper towel to dry the tube at R.T.;(17)D issolve in 30~50 μL ddH2O or TE containing 20 μL/mg DNase-freeRNase A, and incubate at R.T. for 5 min.2.3.6.2Restriction enzyme digestion(1)Add each component following Table 5Table 5 Enzyme digestion of plasmid DNA(2)2.3.6.3Agarose gel electrophoresis(1) Prepare the agarose solution in TAE at the concentration of 0.8%;(2) Prepare a piece of agarose gel;(3) Add 10 μL loading buffer to 20 μL enzyme digestion product;(4) Load the sample as well as the DNA ladder to the slots;(5) Start electrophoresis at 100 V;3Result and Discussion3.1Ethanol precipitationThe gel image is shown in Figure 2Figure 2 Ethanol precipitationLane I: PCR productLane II: pBluescript KSLane M: NEB 1kb DNA ladderCompare Band I and II, they are about the same shade. Considering that the loadingsample of target DNA is 2 μL and vector 1 μL, the concentration ratio of target DNA andvector should be 1:2. During ligation, we add 1 μL vector and 2μL target DNA to maketheir molecule ratio in an appropriate level.3.2TransformationThe gel image is shown in Figure 3Figure 3 IdentificationLane I: Clone ILane II: Clone IILane M: NEB 1kb DNA ladderClone II contains a band at about 0.9 kb, which is expected as a positive clone, whichcan be used in the following process, while clone I is blank in the gel image. It is mainlyand possibly because some mistakes made in the plasmid extraction and purificationsteps.4ConclusionClone II has the recombinant DNA and is proper for the following process.5Reference【1】/。