鲍林规则和价键理论
- 格式:doc
- 大小:29.50 KB
- 文档页数:2


价键理论概述价键理论概述摘要:价键理论是指固体或分⼦中原⼦的价电⼦结构和原⼦与原⼦之间形成的键以及两者关系的理论。
它是从原⼦和原⼦结构层次, 深⼊了解材料⼀种重要理论, 能帮助⼈们设计满⾜需要的新材料。
根据收集到的资料, 对价键理论及其应⽤进⾏扼要地归纳与阐述。
关键词:价键理论共价键键参数⾦属应⽤价键理论起源于1916 年美国科学家G1 N1Lew is[1]提出的电⼦配对理论。
1927 年德国科学家W1 He itler与F1 L London[2]第⼀个⽤量⼦⼒学处理H2分⼦, 揭⽰了共价键的本质。
1930 年前后Pauling[3]和S later[4]等把这个理论发展成为⼀种全⾯的键理论, 称为价键理论。
⾦属的价键理论实质就是⽤电⼦配对法来处理⾦属键。
这⼀理论在⾦属材料中有着重要的指导作⽤, 它能帮助⼈们从电⼦结构和原⼦结构层次了解晶体结构, 并以此寻找需要的⾦属新材料。
因此, 国内外科学家, 在这⽅⾯做了⼤量的⼯作, 鉴于价键理论的重要性, 对其发展与应⽤做扼要的归纳与阐述。
⼀、键价理论的基本知识1.基本概念价键理论是在Pauling 离⼦晶体电价规则基础上发展起来的, 它继承了电价规则中/原⼦的价分配在原⼦所连诸键上0的基本概念, 同时允许原⼦所连诸键的键价做不均匀的分配。
价鍵的主要内容包括以下⼏个⽅⾯:(1)在价键理论或价键法则中, 将在反应中保持不变的最基本的实体称作原⼦。
在由⼴义( Lewis)酸(阳离⼦)与⼴义碱(阴离⼦)组成的离⼦性化合物中, 荷正电者为正价, 荷负电者为负价。
(2)化学计量要求离⼦性(或酸碱)化合物中的总正价与总负价的绝对值相等。
即化合物整体保持电中性的原理。
(3)原⼦以化学键与其近邻原⼦键合, 其键连原⼦数称为该原⼦的配位数, 此数亦为该原⼦参与化学键的成键数。
(4)价键理论认为, 原⼦的价将分配在它所参与的诸键上, 使每个键均有⼀定的键价, 并符合价和规则。


配合物的价键理论配合物中的化学键主要是指配合物内中心离子(或原子)M 与配体L 之间的化学键。
中心离子和配体之间通过什么样的作用力结合在一起?这种结合力的本质是什么?为什么配离子具有一定的空间构型而稳定性又各不相同?19世纪末,维尔纳(Werner A )曾试图回答这些问题,但没有成功。
直到20世纪,在近代原子和分子结构理论建立以后,用现代的价键理论以及晶体场理论、配位场理论和分子轨道理论,才较好地阐明了配合物中化学键的本质。
1931年鲍林首先将分子结构的价键理论应用于配合物,后经他人修正补充,逐步完善成配合物的现代价键理论。
1.配合物价键理论的要点(1) 中心离子(或原子)M 与配体L 形成配合物时,中心离子(或原子)以空的价轨道接受配体中配位原子提供的孤对电子,形成σ配键(用M ←L 表示)。
(2) 中心离子(或原子)所提供的空价轨道必须杂化,与配位原子的充满孤对电子的原子轨道相互重叠,形成配位共价键。
2.中心离子轨道杂化的类型 在配合物的形成过程中,中心离子需提供一定数目的经杂化的能量相同的空的价轨道与配体形成配位键。
中心离子所提供的空轨道的数目,由中心离子的配位数所决定,故中心离子空轨道的杂化类型与配位数有关。
中心离子空轨道的杂化类型除了前面讲过的sp 、sp 2、sp 3杂化外,能量相近的(n-1)d ,n d 轨道也能参与杂化。
(1) 配位数为2的中心离子的杂化类型 讨论[Ag(NH 3)2]+ 配离子的形成 Ag +离子的价电子层结构为: Ag +离子和NH 3形成[Ag(NH 3)2]+配离子时,配位数为2,Ag +需提供二个空轨道。
Ag +离子外层能级相近的一个5s 和一个5p 轨道经杂化,形成二个等价的sp 杂化轨道,容纳二个NH 3中二个配位N 原子提供的二对孤对电子,形成二个配键(虚线内杂化轨道中的共用电子对由配位氮原子提供): 两个sp 杂化轨道在空间成180°,故[Ag(NH 3)2]+配离子的空间构型呈直线形。


价键理论价键理论valence-bond theory,一种获得分子薛定谔方程近似解的处理方法。
又称电子配对法。
历史上最早发展起来的化学键理论。
主要描述分子中的共价键和共价结合,其核心思想是电子配对形成定域化学键。
1产生1927年W.H.海特勒和F.W.伦敦首次完成了氢分子中电子对键的量子力学近似处理,这是近代价键理论的基础。
L.C.鲍林等加以发展,引入杂化轨道概念,综合成价键理论,成功地应用于双原子分子和多原子分子的结构。
价键理论与化学家所熟悉的经典电子对键概念相吻合,一出现就得到迅速发展。
但价键理论计算比较复杂,使得后来发展缓慢。
随着计算技术日益提高,该理论还会有新发展。
1927年,Heitler 和London 用量子力学处理氢气分子H2,解决了两个氢原子之间化学键的本质问题,使共价键理论从典型的Lewis理论发展到今天的现代共价键理论。
海特勒-伦敦方法处理氢分子氢分子的哈密顿算符是:式中rA1、rB1为核A、B与电子1之间的距离;r12为两个电子之间的距离;RAB为两个原子核之间的距离……(图1);1/RAB表示两个原子核之间的势能(氢核和电子电荷皆为1基本电荷单位);1/rA1、1/rB1、…也是势能;墷是拉普拉斯算符。
海特勒-伦敦方法的要点在于如何恰当地选取基态H2的近似波函数Ψ(1,2)(或称尝试波函数),然后用变分公式使氢分子能量E为最低(假定Ψ是归一化的):式中*表示复数共轭。
考虑两个氢原子组成的体系,若两个氢原子A(有电子1)和B(有电子2)的基态波函数为:φA⑴=πexp(-rA1)φB⑵=πexp(-rB2)假如两个氢原子相距很远,那么体系波函数是:Φ1(1,2)=φA⑴φB⑵实际上两个电子是不可区分的。
同样合适的函数是:Φ2(1,2)=φB⑴φA⑵两个函数Φ1和Φ2都对应相同的能量。
海特勒和伦敦就取两个函数的等权线性组合作为H2的变分函数:Ψ(1,2)=c1Φ1+c2Φ2解久期方程得c1=±c2,波函数和能量是:式中s称原子轨道的重叠积分。


化学键理论简介化学键是指将两个或多个原子结合在一起的力,是构成分子和化合物的基本单位。
化学键理论旨在解释化学键形成的原因以及化学键的类型和性质。
本文将介绍几个常见的化学键理论。
1. 价键理论价键理论也称为路易斯理论,是由美国化学家吉尔伯特·路易斯于1916年提出的。
根据这个理论,化学键形成是由于原子之间的电子共享或电子转移。
在化学键中,原子通过共享或转移电子以实现稳定状态。
共价键的形成是通过电子共享形成的,而离子键的形成是通过电子转移形成的。
2. 电子云理论电子云理论也称为量子力学理论,是由奥地利物理学家艾尔温·薛定谔等人在20世纪初提出的。
根据这个理论,电子不能被简单地看作是粒子,而是存在于原子周围的一种云状结构,称为电子云。
在化学键中,电子云之间的重叠是化学键的形成基础。
共价键形成是由于两个原子的电子云的重叠,而离子键形成是由于正负电荷之间的吸引力。
3. 分子轨道理论分子轨道理论是由德国化学家恩斯特·赫尔曼·福克和罗伯特·桥·休伊特于20世纪初提出的。
根据这个理论,分子中的电子不再局限于原子轨道,而是存在于整个分子的分子轨道中。
分子轨道可以是成键轨道(高能级)或反键轨道(低能级)。
共价键的形成是通过成键轨道的重叠,而离子键的形成是通过成键轨道和反键轨道之间的重叠。
4. 杂化轨道理论杂化轨道理论是由美国化学家林纳斯·鲍林在20世纪初提出的。
根据这个理论,原子轨道在形成化学键时会重新组合成一组新的杂化轨道。
杂化轨道具有介于原子轨道之间的性质,可以更好地解释一些分子的形状和键角。
杂化轨道的形成是为了最大限度地重叠,以实现更强的化学键。
5. 价电子对斥力理论价电子对斥力理论也称为VSEPR理论,是由英国化学家罗纳德·吉尔斯彭尼克在1940年代提出的。
根据这个理论,化学键的形成是为了最小化价电子对之间的斥力。
分子的几何形状取决于周围的原子和非键电子对的排列方式。

第二章配合物的化学键理论内容:研究中心原子和配体之间结合力的本性。
目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valence bond theory)由L.Pauling提出一、理论要点:①配体的孤对电子可以进入中心原子的空轨道;中心原子总是用空轨道杂化,然后用杂化轨道接收配体提供的孤对电子。
②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
中心原子的价层电子结构与配体的种类和数目共同决定杂化类型。
③杂化类型决定配合物的空间构型,磁距和相对稳定性。
二、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[ Fe(CN)6]4-sp杂化直线型[AgCl2]-三、内轨型和外轨型若要形成ML6型配合物(L为单齿配体),则需要6个空杂化轨道接收6个L提供的孤电子对,满足该条件的杂化类型有d2sp3和sp3 d2。
尽管这两种杂化都导致八面体型配合物,但前者是次外层(n-1)d轨道,而后者是最外层nd轨道,因此与这两种杂化相应的配合物分别称内轨型和外轨型配合物。
中心原子的价层电子数和配体的性质都是影响配合物内轨型和外轨型的因素。
当d电子数≤3时,该层空d轨道≥2,总是生成内轨型配合物。
当中心原子价层d电子数为7~10时,即使强制d轨道中的电子配对,所能得到的该层空d轨道数也小于2,因此只能用最外层d轨道参与杂化,总是生成外轨型配合物。
当中心原子价层d电子数为4~6时,对于配位能力较强的配体,即配位原子电负性较小,容易给出孤电子对,对中心原子价层d电子排布影响较大,强制d电子配对,空出2个价层d轨道参与d2sp3杂化,生成内轨型配合物.若配体的配位能力较弱,即配位原子电负性较大,则不易给出孤电子对,对中心原子价层d电子排布影响较小,只能用最外层d轨道参与杂化,生成外轨型配合物。

鲍林化学键的本质化学键是指形成化合物的原子之间的连接。
鲍林在1919年提出了化学键的概念,他认为化学键是由原子之间的电子相互作用而形成的。
鲍林认为,原子中的电子围绕在原子核周围,每个电子具有一定的能量和运动状态。
在化学反应中,原子之间的电子会重新排列,形成新的化学键。
鲍林根据电子排布的特点,提出了共价键、离子键和金属键三种化学键的概念,并详细阐述了它们的性质和形成机制。
共价键是指原子间电子的共享。
在共价键中,原子之间的电子通过共用方式共享在双方的轨道上,形成一个共享电子对,将两个原子紧密连接在一起。
共价键的形成需要具备较强的原子间吸引力和一定的电子互斥作用。
共价键的特点是键能较高,键长较短,化合物一般显具有较高的稳定性,同时还具有一定的空间取向性和方向性。
离子键是指带正电荷的金属离子与带负电荷的非金属离子之间的电荷吸引作用。
在离子键中,正负电荷相互吸引,形成离子晶体。
离子键的形成需要具备较强的静电相互作用力和电子转移的能力。
离子键的特点是键能较高,键长较长,化合物一般呈离子晶体结构,具有较高的熔点和溶解度,同时还具有吸湿性和良导电性。
金属键是指金属原子之间的电子云相互重叠形成的键。
在金属键中,金属原子的外层电子云相互重叠,形成一个共享的电子海,使得金属原子紧密连接。
金属键的形成需要具备较低的电子互斥力和较强的金属原子间吸引力。
金属键的特点是键能较低,键长较长,化合物表现出良好的导电性、热传导性和延展性。
鲍林的化学键理论为化学界提供了一种理解化学反应和化合物性质的重要框架。
他的理论揭示了物质世界的基本结构,促进了化学反应和化学合成的发展。
同时,鲍林的化学键理论也为进一步研究和解释其他化学现象提供了思路和方法。
总而言之,鲍林化学键提出了化合物形成的本质是原子之间电子的相互作用,而共价键、离子键和金属键则是电子相互作用不同方式的具体体现。
这一理论为化学界提供了重要的思想工具,推动了化学科学的发展。
杂化轨道理论价键理论简明地阐明了共价键的形成过程和本质,成功解释了共价键的方向性和饱和性,但在解释一些分子的空间结构方面却遇到了困难。
例如CH 4分子的形成,按照价键理论,C 原子只有两个未成对的电子,只能与两个H 原子形成两个共价键,而且键角应该大约为90°。
但这与实验事实不符,因为C 与H 可形成CH 4分子,其空间构型为正四面体,∠HCH = 109.5°。
为了更好地解释多原子分子的实际空间构型和性质,1931年鲍林提出了杂化轨道理论(hybrid orbital theory ),丰富和发展了现代价键理论。
1953年,我国化学家唐敖庆等统一处理了s-p-d-f 轨道杂化,提出了杂化轨道的一般方法,进一步丰富了杂化理论的内容。
1.杂化轨道理论的基本要点杂化轨道理论从电子具有波动性、波可以叠加的观点出发,认为一个原子和其他原子形成分子时,中心原子所用的原子轨道(即波函数)不是原来纯粹的s 轨道或p 轨道,而是若干不同类型、能量相近的原子轨道经叠加混杂、重新分配轨道的能量和调整空间伸展方向,组成了同等数目的能量完全相同的新的原子轨道——杂化轨道(hybrid orbital),以满足化学结合的需要。
这一过程称为原子轨道的杂化(hybridization )。
下面以CH 4分子的形成为例加以说明。
基态C 原子的外层电子构型为2s 22p x 12p y 1。
在与H 原子结合时,2s 上的一个电子被激发到2p z 轨道上,C 原子以激发态2s 12p x 12p y 12p z 1参与化学结合。
当然,电子从2s 激发到2p 上需要能量,但由于可多生成二个共价键,放出更多的能量而得到补偿。
在成键之前,激发态C 原子的四个单电子分占的轨道2s 、2p x 、2p y 、2p z 会互相“混杂”,线性组合成四个新的完全等价的杂化轨道。
此杂化轨道由一个s轨道和三个p 轨道杂化而成,故称为sp 3杂化轨道。
鲍林公式算化合物中离子键的比例
鲍林公式是一个简单、直观的工具,可以帮助我们快速计算化合物
中离子键的比例。
下面我们来详细介绍一下鲍林公式的原理和应用方法。
一、鲍林公式的原理
鲍林公式是由奥地利化学家鲍林于1919年提出的,它基于以下两个假设:
1. 离子键的键能远大于共价键的键能。
2. 化合物中每种离子都是最简单的。
根据这两个假设,鲍林得出了如下公式:
离子键的比例 = (阴离子原子价 + 阳离子原子价)/离子键的键能
其中,阴离子原子价是指阴离子中负电荷的总数,阳离子原子价是指
阳离子中正电荷的总数。
离子键的键能是指形成离子键所需要的能量。
二、鲍林公式的应用方法
下面我们通过一个例子来演示鲍林公式的具体应用方法。
假设要计算氯化钠中离子键的比例,根据鲍林公式,我们需要先计算氯离子和钠离子的原子价和离子键的键能。
氯离子的原子价为1,因为氯原子带有-1的负电荷;
钠离子的原子价为1,因为钠原子带有+1的正电荷;
离子键的键能可以通过查阅资料得知,对于氯化钠而言,离子键的键能为756kJ/mol。
代入鲍林公式,得到:
离子键的比例 = (1+1)/756kJ/mol = 0.0026
也就是说,在氯化钠中,离子键所占的比例非常小,只有0.26%。
三、总结
鲍林公式是一种简便、实用的计算化合物中离子键比例的工具。
但是需要注意的是,此公式只适用于离子性较强的化合物,对于共价化合物并不适用。
在实际应用过程中,还应注意参考其他相关性质,如晶体结构等,进行综合评估。
pauling金属的价键理论简称1.具有自旋相反的未成对电子的两个原子相互接近,可以形成稳定的共价键。
2.原子中未成对的电子数等于原子所能形成的共价键数目。
3.成键电子的电子云重叠越多,核间电子子云密度越大,形成的共价键越牢固。
价键理论,又称电子配对法,其基本要点如下:1.如果a、b两个原子各存有一个磁矩恰好相反的未雄雀的电子,那么这两个固非对电子可以相互接合构成平衡的共价键,这对电子为a、b两原子所共计。
如果a、b各存有两个或三个未雄雀的电子,则磁矩恰好相反的单电子可以两两接合构成共价键或叁键。
如果a原子有两个未成对电子,b原子有一个未成对电子,那么一个a原子能与两个b原子结合形成ab2型分子。
2.共价键就是由成键原子中磁矩恰好相反的固非对电子接合构成的。
一个原子的一个电子和另一个原子的一个电子接合以后,无法再和第二个电子接合。
因为这时其中必存有两个电子的磁矩方向相同而背道而驰。
也就是说一个原子所能够构成共价键的数目就是一定的。
原子中未雄雀的电子数等同于原子所能够构成的共价键数目,这就是共键价的饱和状态性。
医学`教育网搜集整理比如,h原子只有一个固非对电子,它和另一个h原子的固非对电子接合后,就无法再与第二个h原子的电子接合了。
3.共价键的生成是由于自旋相反的单电子相互配对,电子云重叠的结果。
医学`教育网搜集整理因此,当两个原子形成分子时,电子云重叠的程度越大,则两原子间的电子云密度越大,生成的共价键越牢固,所以,在形成共价键时,电子云总是尽可能达到晨大程度的重叠,这叫电子云最大重叠原理。
根据电子云最小重合原理,在构成共价键时,原子间总是尽可能沿着电子云最小重合方向成键。
s电子云呈圆形球形等距原产,p、d、f电子云在空间都存有一定的弯曲方向。
医学`教育网搜集整理在构成共价键时,除了s电子云和s电子云可以在任何方向上都能够达至最小程度的重合外,p、d电子云的重合,只有在一定方向上就可以并使电子云存有最小程度的重合。
鲍林与化学键理论鲍林是世界著名的化学家,他一生涉猎和成就极广,而化学键是他最核心的研究领域。
鲍林在20世纪30年代提出的共振理论、杂化、电负性等化学键理论概念在化学领域深入人心。
1939年,鲍林出版了在科学史上具有划时代意义的《化学键的本质》(The Nature of the Chemical Bond)一书。
这部书彻底改变了人们对化学键的认识,在该书出版后的30年内,共被引用超过16000次。
将化合价(原子的化合能力)的8电子理论总结为路易斯-朗缪尔电子理论,这便是现代电子价键理论的开端。
受前辈学者启迪,鲍林对于原子如何聚集成为分子兴趣大增。
进入加州理工学院攻读物理化学博士学位后,他得到名师指点,应用X 射线进行无机晶体结构的研究,从而掌握了大量关于晶体结构的信息、原子间距离和键角的细节。
在以往的经验及理论观察的基础上,鲍林开始应用量子力学的理论(电子有自旋角动量,具波动特性)来探索化学键的形成与性质,并引入“共振”的思想和“杂化轨道”的概念,用以解释围绕碳原子的4个键的等价性,进而完整地解释了甲烷的正四面体结构。
1931年,他发表了《化学键的本质》等一系列关于化学键研究的论文,名震化学界。
此时,他刚过而立之年。
莱纳斯·卡尔·鲍林(L·C·Pauling ,1901−1994),美国化学家,量子化学和分子生物学的先驱者之一。
1922年获俄勒冈农学院化学工程学士学位,1926年获加州理工学院物理化学博士学位,1933年当选为美国国家科学院院士,1937年任加州理工学院化学与化学工程系主任,1948年被授予总统功勋奖章,1949年任美国化学会主席。
1954年,鲍林因阐明了化学键的本质和分子结构的基本原理,被授予诺贝尔化学奖。
1962年,鲍林又因积极维护世界和平、反对核试验而被授予诺贝尔和平奖。
鲍林是迄今为止世界上唯一两次单独获得诺贝尔奖的科学家,有很高的国际声誉,被誉为人类迄今为止最伟大的20位科学家之一。
鲍林关于估算含氧酸酸性强弱的两条规则是什么?它们对化学研究有何指导价值?鲍林提出两条规则用以粗略估算含氧酸O p E(OH)q的p K a○一值, 含氧酸通式中的p 和q分别代表末端氧原子和羟基的数目。
1. 电中性氧合酸的p K a○一≈8-5p。
即p=0时p K a○一≈8;p=1时p K a○一≈3;p=2时p K a○一≈-2。
2. 多元酸(q>1)多步质子转移反应的p K a○一值逐级增加5。
例如硫酸(O)2S(OH)2(p=2, q=2)的p K a1○一≈-2,而p K a2○一≈+3。
这些规则的成功可由表1提供的实例得到支持,估算值与实验值之间的误差仅约±1。
实际工作中通常不要求用鲍林规则得到p K a○一值,因为与其这样, 倒不如从化学手册中去查找。
鲍林规则也不能用来解释化学现象,而规则本身倒是需要作解释。
例如, 根据鲍林规则, 一个具体酸的强弱似乎与通式中的E无关, 这在化学上是难以理解的。
表1 鲍林规则应用选例鲍林规则最有趣的用途也许是发现结构异常现象, 如果实验值与估算值差异太大, 则可能是结构异常的一种信息。
例如H2CO3, 估算的p K a○一为3, 显著偏离了实验值6.4。
后来研究证实, 这种偏离是因为处理实验数据时直接将溶于水中的CO2浓度当作H2CO3浓度,而实际溶解的CO2大约只有1%转化为H2CO3。
如果采用H2CO3的实际浓度,算得的数值约为3.6, 与估算值的误差在±1之内。
另外的例子有如亚磷酸,实验值更接近于表中p=1的那个估算值。
这一事实暗示分子中存在一个末端氧原子。
根据亚磷酸的化学式H3PO3,似乎不存在末端氧原子[P(OH)3], 容易给人造成错觉以为它是个三元酸。
研究表明, 亚磷酸实际上是个二元酸,其中含有一个末端氧原子和一个与磷原子直接键合的氢原子。
1。
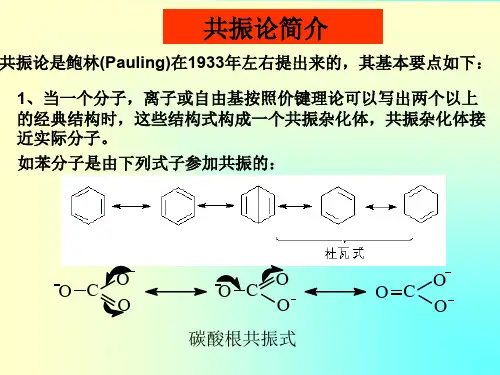
鲍林规则和价键理论
1928年,鲍林根据当时已测定的晶体结构数据和晶格能公式所反映的关系,提出了判断离子化合物结构稳定性的规则──鲍林规则。
鲍林规则共包括五条规则:
鲍林第一规则:在离子晶体中,在正离子周围形成一个负离子多面体,正负离子之间的距离取决于离子半径之和,正离子的配位数取决于离子半径比。
鲍林第二规则:在一个稳定的离子晶体结构中,每一个负离子电荷数等于或近似等于相邻正离子分配给这个负离子的静电键强度的总和,其偏差≤1/4价。
鲍林第三规则:在一个配位结构中,共用棱,特别是共用面的存在会降低这个结构的稳定性。
其中高电价,低配位的正离子的这种效应更为明显。
鲍林第四规则:若晶体结构中含有一种以上的正离子,则高电价、低配位的多面体之间有尽可能彼此互不连接的趋势。
鲍林第五规则:在同一晶体中,组成不同的结构基元的数目趋向于最少。
鲍林(Pauling)规则是根据离子晶体的晶体化学原理,通过对一些较简单的离子晶体结构进行分析,总结归纳出的五条规则。
氧化物晶体及硅酸盐晶体大都含有一定成分的离子键,因此,在一定程度上可以根据鲍林规则来判断晶体结构的稳定性。
第一规则实际上是对晶体结构的直观描述,如NaCl晶体是由[NaCl6]八面体以共棱方式连接而成。
利用第二规则可以判断晶体是否稳定,同时也可以判断共用一个顶点的多面体的数目。
例如,在CaTiO3结构中,Ca2+、Ti4+、O2-离子的配位数分别为12、6、6。
O2-离子的配位多面体是[OCa4Ti2],则O2-离子的电荷数,与O2-离子的电价相等,故晶体结构是稳定的。
又如,一个[SiO4]四面体顶点的O2-离子还可以和另一个[SiO4]四面体相连接(2个配位多面体共用一个顶点),或者和另外3个[MgO6]八面体相连接(4个配位多面体共用一个顶点),这样可使O2-离子电价饱和。
第三规则又称为多面体共顶、共棱、共面规则。
两个配位多面体连接时,随着共用顶点数目的增加,中心阳离子之间距离缩短,库仑斥力增大,结构稳定性降低。
第四规则又称为不同配位多面体连接规则。
第五规则这个规则关注与晶体结构的周期性和对称性,如果组成不同的结构基元较多,每一种基元要形成各自的周期性、规则性,则它们之间会相互干扰,不利于形成晶体结构。
价键理论(valence-bond theory),是在鲍林规则的基础上发展起来的。
是一种获得分子薛定谔方程近似解的处理方法。
又称电子配对法。
主要描述分子中的共价键和共价结合,其核心思想是电子配对形成定域化学键。
其包含了共价键的形成、方向性和饱和性、共价键的键型。
电价规则是鲍林五个规则的核心。
它可表述为:在一个稳定的离子化合物结构中,每一负离子的电价等于或近似等于从邻近的离子至该负离子的各静电键强度的总和。
这一规则的物理基础在于如在结构中正电位较高位置安放电价较高的负离子时,给构会趋于稳定,而某一正离子至该负离子的静电键的强度,正是有关正离子在该处所引起正电位的量度。
近几年结合鲍林规则利用价键理论对晶体的结构及性质的研究有了突破行的进展。
就铌酸锂晶体而言,它是一种典型的非化学计量比多功能晶体材料。
之前,我们多是利用能量计算、实验分析讨论晶体的结构。
但是价键理论分析,引入化学势,通过键长、将强等的分析,不但使是我们假设的晶体结构具体话,也从化学键的角度讨论了晶体内部结构对晶体性能的影响。
所以鲍林规则和价键理论为材料的学特备是晶体学的研究提供了一个更方便的平台,促进了材料学研究突破性的进展。