第二章 红外光谱1
- 格式:doc
- 大小:453.00 KB
- 文档页数:11










第二章 红外光谱(高校教师研究生班)红外光谱(IR):一种分子吸收光谱,又称为分子的振—转光谱。
当用连续波长的红外光照射某一物质时,该物质就吸收一定波长的红外光的光能,并将被吸收的光能转化为分子的振动和转动能。
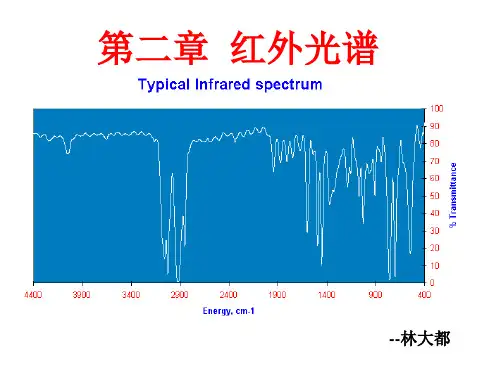
以波长为横坐标,百分吸光率或百分透过率为纵坐标把谱图记录下来,就可以得到该物质的红外光谱。
表2.1 红外光谱的分类大多数的有机化合物和许多无机化合物的化学键的振动基频都出现在中红外区。
中红外的吸收光谱在化合物的构造解析和定量分析方面有重要的意义,所以中红外是我们研究的主要对象。
用频率表示红外光谱不方便,通常用波数〔cm -1〕表示,波数与波长的关系为:波数[cm -1] =波长410 波长的单位是微米(µm); 1cm=104µm 即1 cm 内有多少个波数2.1 根本原理2.1.1 分子的振转光谱分子总是在不断地运动,各种运动状态都有一定的能态。
分子的总能量为:E = E 0+ E 平 + E 转 + E 振 + E 电E 0—分子的内能,其能量不随分子的运动而改变。
E 平——是温度的函数,平动时不产生偶极矩的变化,无红外吸收。
所以红外光谱只与分子的转动能、振动能和分子内电子的运动能有关。
分子可以吸收红外光的辐射能从低能态跳到高能态,但不是任意的,必须满足量子化条件〔选律〕。
不管是振动能还是转动能,都有能级差〔 〕,只有当辐射光光子的能量正好等于两能级间的能量差时,分子才能吸收辐射能从低能级跳到高能级,产生吸收光谱。
振动能级的= 0.5~1 eV, 中红外光的辐射能正好在此X 围。
当振动发生时总伴随有转动能级产生,所以,IR 光谱称为振—转光谱。
2.1.2 双原子光谱的振动光谱对于双原子分子(以HCl 为例),可看把两个原子看成质量不等的两个小球m 1和m 2 ,把它们的化学键类比为质量可以不计的弹环,两原子在平衡位置的振动可以近似为简谐振动,根据虎克定律,简谐振动的频率为: μκπυ21= 〔2.1〕 用波数(cm -1)表示为: υ〔cm -1〕=μκπc 21 〔2.2〕 E E m 12m式中: C —光 速κ—力常数 [单键约为5×102 N ·m -1;双键和三键是单键的2倍或3倍]μ—折合质量:μ= m 1·m 2 / m 1+ m 2 [ m 1、m 2 是两个原子的质量〔克〕]:130421=c π; κ=5.1 〔毫达因/埃〕; 972.05.3515.351=+⨯=μ〔HCl 〕 根据[2.2]式: υ(cm -1) = 1304972.01.5=2990 [cm-1] 实际测得HCl 分子的H 、Cl 伸缩振动频率为2886cm -1。
第二章红外光谱(Infrared Spectroscopy,IR)和拉曼光谱(Raman Spectroscopy)•用于检测特殊的官能团(Functional Groups)。
•不能给出整个分子结构(Total Structures)和片断关联(Connectivity)的信息。
•红外光谱是分子吸收光谱,涉及偶极矩改变的分子振动和转动能级跃迁。
一个分子吸收能量后,它的能量变化总和△E为,即式中△Ee最大,一般在1~20eV之间。
分子的振动能级变化△Ev大约比电子运动能量变化△Ee小10倍,一般在0.05~leV之间。
分子的转动能级变化△Er大约比分子的振动能级变化△Ev小10倍或100倍,一般小于0.05eV。
分子的振动能量比转动能量大,当发生振动能级跃迁时,不可避免地伴随有转动能级的跃迁,所以无法测量纯粹的振动光谱,而只能得到分子的振动-转动光谱,这种光谱称为红外吸收光谱。
2.1.2 红外光谱的产生满足两个条件:(1)红外光波的频率和分子中基团的振动频率一致。
分子吸收红外辐射后,由基态振动能级(ν=0)跃迁至第一振动激发态(ν=1)时,所产生的吸收峰称为基频峰。
在红外吸收光谱上除基频峰外,还有振动能级由基态(ν=0)跃迁至第二激发态(ν=2)、第三激发态(ν=3)…,所产生的吸收峰称为倍频峰。
由ν= 0跃迁至ν= 2时,吸收的红外谱线是分子振动频率的二倍,产生的吸收峰称为二倍频峰。
由ν= 0跃迁至ν= 3时,吸收的红外谱线是分子振动频率的三倍,产生的吸收峰称为三倍频峰。
其它类推。
在倍频峰中,二倍频峰还比较强。
三倍频峰以上,因跃迁几率很小,一般都很弱,常常不能测到。
由于分子非谐振性质,各倍频峰并非正好是基频峰的整数倍,而是略小一些。
以HCl为例:基频峰(ν0→1)2885.9 cm-1最强二倍频峰(ν0→2 )5668.0 cm-1较弱三倍频峰(ν0→3 )8346.9 cm-1很弱四倍频峰(ν0→4 )10923.1 cm-1极弱五倍频峰(ν0→5 )13396.5 cm-1极弱除此之外,还有合频峰(ν1+ν2,2ν1+ν2,…),差频峰(ν1-ν2,2ν1-ν2,…)等,这些峰多数很弱,一般不容易辨认。
第二章 红外光谱(高校教师研究生班)红外光谱(IR):一种分子吸收光谱,又称为分子的振—转光谱。
当用连续波长的红外光照射某一物质时,该物质就吸收一定波长的红外光的光能,并将被吸收的光能转化为分子的振动和转动能。
以波长为横坐标,百分吸光率或百分透过率为纵坐标把谱图记录下来,就可以得到该物质的红外光谱。
表2.1 红外光谱的分类大多数的有机化合物和许多无机化合物的化学键的振动基频都出现在中红外区。
中红外的吸收光谱在化合物的结构解析和定量分析方面有重要的意义,所以中红外是我们研究的主要对象。
用频率表示红外光谱不方便,通常用波数(cm -1)表示,波数与波长的关系为:波数[cm -1] =波长410波长的单位是微米(µm); 1cm =104 µm 即1 cm 内有多少个波数2.1 基本原理2.1.1 分子的振转光谱分子总是在不断地运动,各种运动状态都有一定的能态。
分子的总能量为:E = E 0+ E 平 + E 转 + E 振 + E 电 E 0—分子的内能,其能量不随分子的运动而改变。
E 平——是温度的函数,平动时不产生偶极矩的变化,无红外吸收。
所以红外光谱只与分子的转动能、振动能和分子内电子的运动能有关。
分子可以吸收红外光的辐射能从低能态跳到高能态,但不是任意的,必须满足量子化条件(选律)。
不论是振动能还是转动能,都有能级差( ),只有当辐射光光子的能量正好等于两能级间的能量差时,分子才能吸收辐射能从低能级跳到高能级,产生吸收光谱。
振动能级的 = 0.5~1 eV , 中红外光的辐射能正好在此范围。
当振动发生时总伴随有转动能级产生,所以,IR 光谱称为振—转光谱。
2.1.2 双原子光谱的振动光谱对于双原子分子(以HCl 为例),可看把两个原子看成质量不等的两个 小球m 1和m 2 ,把它们的化学键类比为 质量可以不计的弹环,两原子在平衡位置 的振动可以近似为简谐振动,根据虎克定 律,简谐振动的频率为:μκπυ21=(2.1) 用波数(cm -1)表示为: υ(cm -1)=μκπc 21 (2.2)式中: C —光 速κ—力常数 [单键约为5×102 N ·m -1;双键和三键是单键的2倍或3倍] μ—折合质量:μ= m 1·m 2 / m 1+ m 2 [ m 1、m 2 是两个原子的质量(克)]已知:130421=c π; κ=5.1 (毫达因/埃); 972.05.3515.351=+⨯=μ(HCl )根据[2.2]式: υ(cm -1) = 1304972.01.5=2990 [cm-1] 实际测得HCl 分子的H 、Cl 伸缩振动频率为2886cm -1。
表明:把双原子E E m 12m分子示作偕振子,利用经典力学讨论双原子分子的振动,能基本说明分子振动光谱的主要特征。
可以粗略地计算双原子分子或多原子分子中双原子化学键的振动频率。
例1:计算 C=O 双键的伸缩振动频率。
已知k=12.06;μ=6.85υ(cm -1) = 130485.606.12=1724 [cm-1]例2:计算 CN 三键的伸缩振动频率。
已知k=15;μ=6.46υ(cm -1) = 130446.615=1987 [cm-1]2.1.3 多原子分子的振动光谱由于多原子分子有多个原子,加之排列方式的相互影响,使光谱变得十分复杂。
但也正是由于这中谱图的复杂性才包纳了大量的结构性息。
1、 振动型式的表示伸缩振动(υ) — 键长变化,键角不变化。
伸缩振动(υ): υas 不对称伸缩振动(反对称伸缩振动) υs 对称伸缩振动特 征: 双原子分子只有一种伸缩振动形式。
当一个中心原子与两个以上相同原子相连时,就会有不对称与对称伸缩振动。
弯曲振动:原子垂直于价键的振动,键长不变,键角发生变化,又称 变形振动。
弯曲振动有如下几种具体的振动形式:δ: 剪式振动(面内变形振动) γ: 面内摇摆振动ω: 面外摇摆振动(面外变形振动)τ:扭曲振动δs对称变形(弯曲)振动δas 不对称变形(弯曲)振动以甲基、亚甲基为例—CH3:νS:对称伸缩振动:2872cm-1νas:不对称伸缩振动:2962cm-1δS:对称弯曲振动:1380cm-1δas:不对称弯曲振动:1460cm-1—CH2:νS:对称伸缩振动:2853cm-1νas:不对称伸缩振动:2926cm-1δ:剪式振动(面内变形振动):1465cm-1γ:面内摇摆振动:720cmω:面外摇摆振动:1300cm-1-1τ:扭曲振动:1250c2 基本振动数基本振动—简正振动,又称振动自由度N个原子组成的分子有3N个自由度。
分子本身从整体考虑就有三个平动自由度和3个转动自由度(线性分子为2个),所以分子的振动自由度为:3N-6个自由度(线性分子为3N-5)。
例如:HCl 分子振动自由度=3×2-5 = 1H2O 分子振动自由度=3×3-6=3分子有多少个振动自由度就有多少个简正振动数,就有多少个简正振动频率。
多原子分子的红外光谱有时很复杂,除了基频以外〈简正振动频率〉还可以出现倍频、组频、差频吸收带。
〈峰较弱〉倍频:为基频的倍数。
〈2ν1,2ν2…〉差频:两个频率的差〈ν1-ν2,ν3-ν4…〉组频:两个或多个频率之和〈ν1+ν2,ν1+ν3…〉虽然这些峰强度较弱,但有时可以反映出某些结构特征。
(取代苯在2000—1600cm-1的谱带)3选律和对称性通常,基频谱带的数目总是少于理论数例如:CO2有四个自由度〈四种振动形式〉,而实际上只有二个红外吸收谱带,这与分子能级跃迁的选律和分子的对称性有关。
量子力学和实验结果证明:只有偶极矩发生变化的振动才有红外吸收,这种振动为红外活性振动。
红外吸收的实质是振动分子与红外辐射发生能量交换的结果。
当两个原子组成的化学键在键轴上振动时,伴有偶极矩的变化,电荷分布发生变化,产生瞬间变化的电磁场,当这个交变磁场的频率正好等于红外辐射光的频率时,就会产生红外吸收。
具有永久性偶极矩的分子或化学键总是有红外吸收的。
如:NO ,HCl ,C=O …能产生瞬间偶极矩的分子或化学键也有红外吸收。
如CO 2没有偶极矩变化的分子不吸收红外辐射。
如H 2,N 2,O 2对称取代的化学键在振动时没有偶极矩的变化,也观察不到红外吸收光谱。
简并:吸收频率相同的两个谱带重迭的现象。
引起谱带减少的原因之一。
对于有重复结构单元的高分子,大分子易引起谱带的重迭而发生简并。
2、 吸收谱带和强度化学键的极性愈大,振动时偶极矩变化愈大,吸收谱带(的强度)越强。
C=O ,SiO ,C -Cl 有很强的IR 吸收带 C=C ,C -C ,C -N 其伸缩振动吸收带很弱。
分子对称性愈高,振动时偶极矩变化越小,吸收谱带越弱。
三氯乙烯 四氯乙烯 三氯乙烯在ν(C=C )1585cm-1有吸收带,而四氯乙烯由于(全对称),C=C 键是全对称的伸缩,所以在1585cm -1处观察不到吸收带。
(但有拉曼光谱)吸收带强弱的表示方法: 很强:VS 强: S 中: m 弱: w 宽: b2.2 基本特征频率分组2 . 2.1 第一组:3650~2500 cm-1特征基团振动形式范围(cm-1) OH υO-H3650~3200COOH υO-H3560~2500NH2,NH υN-H3500~3200=C-H υ=C-H 3100~3000C-H υC-H3000~2840 2. 2. 2 第二组:2300~1900 cm-1特征基团振动形式范围(cm-1) 炔基R-C≡C-H υC≡C 2160~2120R-C≡C-R'υC≡C 2050~2000R-C≡C-R ——无吸收带腈基R-C≡N υC≡N2270~2200积累双键:在此范围有强的不对称与对称伸缩振动吸收(亚胺) N=C=N υ N=C=N 2160~2120 (烯亚胺) C=C=N υ C=C=N 2050~2000 (重氮化合物) C=N=N υ C=N=N 2132~2012 (叠氮化合物) N=N-N υ N=N=N2160~2120 (异氰酸酯) N=C=O υ N=C=O2280~2250 (异硫氰酸酯) N=C=S υ N=C=S2100左右(丙二烯) C=C=C υ C=C=C1950左右C ORR ClR ClCl FF C OC OC OCH 2CH 2C OCH CH CH C O2. 2. 3 第三组: 1900~1500 cm -1特征基团 振动形式 范围(cm -1)烯 键 C=C υ C=C 1680~1620 苯环骨架 C=C υ C=C 1600~1500 亚胺,吡啶类 C=N υ C=N 1680~1500 羰 基 C=O υ C=O 1900~1600羰基在此范围内有强吸收带,干扰较少易于识别。
受不同基团的影响,吸收波数有明显的差别,包含了许多的结构信息。
影响羰基吸收峰位移的主要因素有:(1) 诱导效应—吸电子作用使得C=O 双键性增强,力常数变大,引起吸收频率向高波数移动。
取代基的电负性越强,或取代基的数目越多,向高波数方向位移的值也越大。
以丙酮的C=O 吸收频率(1715cm -1)作参考标准: 基 团υ C=O 1715 1802 1828 1928(2) 共轭效应π—π共轭—当C=O 与不饱和的双键、三键、苯环共轭时,形成大π键,电子云密度趋于平均化,导致C=O 的力常数减小,吸收峰向低波数方向移动。
例:1725~17051685~1665CH CH CH CH C OC OR CO2C O RC OR ..O R COR.CH CH2COR1670~16601700~16801670~1660P —π共轭—具有孤对电子的原子与C=O 共轭。
孤对电子向羰基上偏移,羰基上的电子云又向氧原子上偏移,使羰基的双键性减弱,力常数减小,吸收频率向低波数移动。
由于N 原子上的P 电子(孤对电子)与C=O 共轭,导致酰胺C=O 的吸收波数(1680 cm -1)明显地低于一般羰基(1715 cm -1)。
注 意:在一个化合物的结构之内,可能存在多种作用的同时影响。
酯羰基比酮羰基具有更大的吸收波数是因为O 原子即提供孤对电子又具有很强的电负性。
在此,诱导效应起到了主导作用。
υ C=O :~1735 cm -1醋酸乙烯酯、醋酸苯酯比一般的酯C=O 高出约35个波数,其原因为两个相反的共轭结构作用力互相抵消,此时诱导效应起到了支配作用。
υ C=O :1776 cm -1 υ C=O :1770 cm -1R O RC R O RC O OOOOO OOOO NHONHONHO ~cm11800~cm11775~cm 11745~cm11710~cm11820~cm11770~cm11735~cm11745~cm11705~cm11665 (3) 环的张力—张力越大,羰基的吸收频率(波数)也越大。