转录组测序结题报告
- 格式:pdf
- 大小:987.49 KB
- 文档页数:11

转录组测序结题报告转录组测序结题报告篇一:转录组测序问题集锦转录组测序问题集锦转录组是某个物种或者特定细胞类型产生的所有转录本的集合,转录组测序(RNA-seq) 是最近发展起来的利用深度测序技术进行转录分析的方法,可以对全转录组进行系统的研究。
Roche GS FLX Titanium 、Illumina Solexa GA IIx和AB SOLID 4均可以对转录组进行测序,Roche GS FLX Titanium与Illumina Solexa GA IIx和AB SOLID 4相比,拥有更长的读长和较小的数据量,适用于表达量较高基因的RNA全长测序。
但是对低表达丰度的基因,可能需要多次测序才能得到足够的数据,成本比较高,而Illumina Solexa GA IIx和AB SOLID 4数据读取量大,能够得到较高的覆盖率,可以较好的降低成本。
若是位置基因组序列的物种,则Roche GS FLX Titanium测序更有优势,其较长的读长便于拼接,获得更好的转录本数据。
转录组测序可以供研究者在转录本结构研究(基因边界鉴定、可变剪切研究等),转录本变异研究(如基因融合、编码区SNP研究),非编码区域功能研究(Non-coding RNA研究、miRNA前体研究等),基因表达水平研究以及全新转录本发现等方面进行深入研究。
研究转录组的方法有哪些?目前研究转录组的方法主要三种,基于杂交技术的cDNA 芯片和寡聚核苷酸芯片,基于sanger测序法的SAGE (serial analysis of gene expression)、LongSAGE和MPSS(massively parallel signature sequencing),基于第二代测序技术的转录组测序,又称为RNA-Seq。
转录组测序比其他研究方法有哪些优势?(1)可以直接测定每个转录本片段序列、单核苷酸分辨率的准确度,同时不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题;(2)灵敏度高,可以检测细胞中少至几个拷贝的稀有转录本;(3)可以对任意物种进行全基因组分析,无需预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行转录组分析,同时能够检测未知基因,发现新的转录本,并准确地识别可变剪切位点及cSNP,UTR区域。

转录组分析报告介绍转录组分析是研究基因组中转录过程的研究领域。
通过转录组分析,我们可以了解到在特定条件下细胞中正在转录的所有基因。
这些信息对于理解细胞功能、疾病发展以及生物技术的开发都非常重要。
本报告将介绍转录组分析的一般步骤和常用方法。
步骤一:实验设计转录组分析的第一步是设计实验。
在这个步骤中,我们需要确定要研究的样本类型、实验条件和重复次数。
合理的实验设计可以最大程度地减少误差,并提高结果的可靠性。
步骤二:RNA提取在转录组分析中,我们需要从样本中提取RNA。
RNA是细胞中转录的产物,它可以反映细胞中正在表达的基因信息。
RNA提取的质量和纯度对后续的转录组分析非常重要。
常用的提取方法包括酚氯仿法、磁珠法和硅胶膜法等。
步骤三:RNA测序RNA测序是转录组分析的核心步骤之一。
通过RNA测序,我们可以将RNA样本转化为对应的DNA序列,并确定每个基因的表达水平。
常见的RNA测序技术包括Sanger测序、二代测序和三代测序等。
二代测序技术如Illumina和Ion Torrent等已经成为转录组分析的主流技术。
步骤四:数据预处理RNA测序会产生大量的原始数据,这些数据需要进行预处理以去除噪音和提高数据质量。
数据预处理包括去除低质量的reads、去除接头序列、去除重复序列和过滤低表达基因等。
预处理后的数据可以为后续的分析提供可靠的基础。
步骤五:差异表达基因分析差异表达基因分析是转录组分析的重要环节之一。
通过比较不同条件下基因的表达水平,我们可以找到与特定条件相关的差异表达基因。
常用的差异表达基因分析方法包括DESeq、edgeR和limma等。
这些方法可以帮助我们发现与特定条件相关的生物学过程和信号通路。
步骤六:功能注释和富集分析一旦确定了差异表达基因,我们可以对这些基因进行功能注释和富集分析。
功能注释可以帮助我们了解差异表达基因的功能和参与的生物学过程。
而富集分析可以帮助我们发现差异表达基因在特定功能和通路中的富集情况。

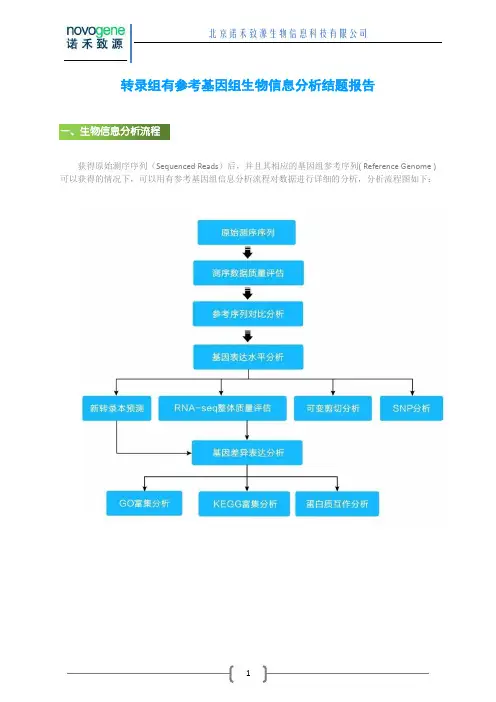
转录组有参考基因组生物信息分析结题报告获得原始测序序列(Sequenced Reads)后,并且其相应的基因组参考序列( Reference Genome )可以获得的情况下,可以用有参考基因组信息分析流程对数据进行详细的分析,分析流程图如下:1. 原始序列数据高通量测序(如Illunima HiSeq TM2000/ Miseq等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
测序样品中真实数据随机截取结果如下:@HWI-ST1106:227:D14F6ACXX:1:1101:1202:2188 1:N:0:GCCAAT CGGATGATCTTCTTAATCTCTCCTTGCATAGTTATGAAACAGTCCGTGGACTTGCTGGAAAATCTCTCTTGAAGATGATGAAGAGATGGCCCTCTACAAT +CCCFFFDFFHHHHJJJJJIJIGGGIGICIGIIJEIIJIIJJI@DHEDHECFGGAHGGJGHIICGEEIEHGGGIECEEHH@HE>C@EBBE@CCDDCCCDDC @HWI-ST1106:227:D14F6ACXX:1:1101:1237:2217 1:N:0:GCCAAT GAAGGTGAGTCTGAGGAGGCCAAGGAGGGAATGTTTGTGAAAGGATATGTCTACTAAGATATTAGAAAGTATGTACTACTACTACTACTACATGTTTTCA +@@@FDADDFDHFHIIIDHIIJJJGICGGGCGHGFIGHBHEHHGI;BDHHCFGCHIIIIEHGIGHHIJJE7??ACHCDFFFFFEEECCEE>C>ACCCDC>@ @HWI-ST1106:227:D14F6ACXX:1:1101:1382:2195 1:N:0:GCCAAT TTTTGCAACAATGGCTTCCACCATGATGACTACTCTACCACAGTTCAATGGACTCAAACCCCAACCTTTCTCAGCTTCTCCAATTCAAGGCTTGGTGGCA +@@@DD3DDFFFF:CDGI@GIEEDH<F49C?EGFBF9?FF?C@BFEFGIII3BDDFFIIG7FFFIIBEFFIFDC3ACBDDDBD@>@AAD;;;@@####### @HWI-ST1106:227:D14F6ACXX:1:1101:1255:2239 1:N:0:GCCAAT CGGATTTTCAAGGGCCGCCGGGAGCGCACCGGACACCACGCGACGTGCGGTGCTCTTCCAGCCGCTGGACCCTACCTCCGGCTGAGCCGATTCCAGGGTG +CCCDFFFFHHH?FHIIIJJJJJIGBEHHJJBHBDDCDAC??@@BDBBBBD8BDDCDDACC@A?@BBB@<<CB?CB<AD?9<B@>(8>?395?4:(:<@## @HWI-ST1106:227:D14F6ACXX:1:1101:1423:2239 1:N:0:GCCAAT CTTGTATTGCTCTCCCACAACCCCGTTTTCACGGTTTAGGCTGCTCCCATTTCGCTCGCCGCTACTACGGGAATCGCTTTTGCTTTCTTTTCCTCTGGCT +CCCFDFFFHHHHHJJIJJJJJIJJGGIHIIGIIJGIGGIJJGGGJGIJ>FGIIGHGGBEHBCCBBDDD@BB@@<AABDDBCACDCDACDCD@:>@C::@C2.测序数据质量评估2.1 测序错误率分布检查测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。

转录组结题报告一、引言转录组研究是生物科学领域中的重要研究方向之一,其对于基因表达调控、疾病发生机制等方面的理解具有重要意义。
本课题旨在探究某种生物在特定条件下的转录组表达谱,以期为理解其基因表达调控机制提供依据。
二、方法1. 实验材料本实验选取了某种生物在特定条件下的多个组织样本,包括健康组织、病变组织以及药物处理后的组织等。
2. 实验方法(1)RNA提取:采用Trizol法提取样本中的总RNA。
(2)建库:将RNA进行逆转录,构建测序文库。
(3)测序:使用Illumina测序平台进行测序,获取原始数据。
(4)数据分析:对原始数据进行质量控制和数据分析,包括基因表达量、差异表达基因分析等。
三、结果1. 基因表达谱通过对测序数据进行质量控制和数据分析,我们获得了每个样本的基因表达谱。
结果显示,在不同样本中,基因表达水平存在显著差异。
其中,一些基因在特定组织中高表达,而在其他组织中低表达,这些基因可能参与了该组织的特定生物学过程。
2. 差异表达基因分析为了进一步理解基因表达调控机制,我们对不同样本之间的基因表达水平进行了差异表达分析。
结果显示,在健康组织和病变组织之间,有数百个基因的表达水平存在显著差异。
这些基因可能参与了疾病的发生和发展过程。
此外,我们还发现一些基因在药物处理后的表达水平发生了显著变化,表明这些基因可能对药物反应具有潜在影响。
四、讨论本实验通过转录组测序技术,获得了某种生物在特定条件下的转录组表达谱。
通过对表达谱的分析,我们发现了一些可能与疾病发生、药物反应相关的基因。
然而,这些发现仍需进一步验证和深入研究。
例如,可以进一步研究这些基因的表达调控机制、与疾病的关系以及潜在的治疗靶点等。
此外,随着新一代测序技术的不断发展,我们可以更深入地研究转录组学领域的其他问题,如转录本结构、可变剪切等。
五、结论本课题通过转录组测序技术,探究了某种生物在特定条件下的转录组表达谱。
实验结果表明,该生物的基因表达水平在不同样本中存在显著差异,这些差异可能与疾病发生、药物反应等相关。


重测序结题报告1. 引言重测序是对DNA或RNA序列进行高通量测序的过程。
通过重测序,我们可以获取组织或个体的基因组或转录组信息,并对基因型、表达水平、基因结构等进行分析。
本文档旨在总结重测序实验的设计、数据分析方法和结果,并对实验的可行性和准确性进行评估。
2. 实验设计2.1 样本选择与准备在本次实验中,我们选择了10个病人的癌细胞样本和10个正常对照组的非癌细胞样本。
样本采集和处理过程遵循了严格的操作规范,并确保了样本的纯度和完整性。
2.2 文库构建和测序使用Illumina HiSeq X10高通量测序平台进行测序。
首先,将样本DNA进行库构建,包括DNA片段化、末端修复、连接接头、PCR扩增等步骤。
然后,将文库进行定量和质量检测,确保文库的质量和浓度符合要求。
最后,将文库进行测序,生成原始测序数据。
3. 数据分析3.1 数据质控对原始测序数据进行质量控制,包括去除接头序列、低质量序列和含有N碱基的序列。
使用FastQC和Trimmomatic等工具对数据进行过滤和修剪。
经过质控后,得到高质量的测序数据,用于后续分析。
3.2 数据比对将测序数据与参考基因组进行比对,以确定序列的来源和定位。
常用的比对工具有Bowtie、BWA和STAR等。
根据比对结果,可以得到每个样本的比对率和覆盖度等信息。
3.3 变异检测通过比对结果,对样本中存在的SNP、InDel和结构变异等进行检测。
常用的变异检测工具有GATK、SAMtools和FreeBayes等。
通过统计和分析得到的变异信息,可以评估样本的基因型和变异频率等。
3.4 差异表达分析对转录组数据进行差异表达分析,以确定基因在癌细胞和正常对照组间的差异表达。
常用的差异表达分析工具有DESeq2、edgeR和limma等。
通过统计和分析得到的差异表达基因,可以进一步研究其功能和调控网络。
4. 结果与讨论实验中,我们成功完成了10个病人的癌细胞样本和10个正常对照组的非癌细胞样本的重测序。

转录组分析报告1. 引言转录组是一组特定生物体细胞或组织中主动转录的所有RNA分子的总和。
转录组分析是通过高通量测序技术,如RNA-seq等,研究生物体在特定生理或病理状态下的基因表达模式和转录水平的变化。
转录组分析在基因功能研究、疾病机制解析和新药研发等领域具有重要应用价值。
2. 实验设计本次实验旨在分析转录组在不同处理条件下的差异表达基因。
我们选取了A和B两个处理组进行对比分析。
每个组别包含3个重复样本,共计6个样本。
样本采集后,我们使用RNA提取试剂盒提取转录组RNA,然后使用Illumina HiSeq平台进行RNA-seq测序。
3. 数据处理3.1 数据质控首先对测序数据进行质量控制,使用FastQC软件分析测序数据的质量分数和碱基分布。
结果显示,测序数据质量良好,无需进行过滤或修剪操作。
3.2 数据预处理在数据预处理过程中,我们主要进行了以下步骤: 1. 使用Bowtie2软件将测序数据比对到参考基因组;bowtie2 -x reference_genome -U input_fastq -S output_sam2.使用Samtools软件将比对结果转换为BAM格式;samtools view -S -b input_sam > output_bam3.使用StringTie软件进行转录本拼接和定量分析;stringtie -G annotation_file -o output_gtf input_bam经过数据预处理后,我们获得了每个基因的表达计数和转录本的FPKM值。
4. 差异表达分析利用DESeq2软件对处理组A和B的差异表达基因进行分析。
在进行差异表达分析之前,我们首先进行了归一化处理,通过计算基因的大小因子来消除测序深度和基因长度之间的偏差。
然后,对处理组A和B之间的基因表达差异进行了t检验,并进行了多重检验校正。
最终,我们选择了在p值<0.05和|log2(fold change)|>1的条件下,认定差异表达基因具有统计学意义。

测序报告总结与反思范文引言测序报告是进行DNA测序后所生成的结果展示和分析的文档,通过阅读报告可以获得关于DNA序列的各种信息。
本文将对测序报告进行总结与反思,总结测序过程中的经验和教训,反思测序结果的可靠性和局限性,以期提高测序的质量和效果。
总结测序过程测序过程中,我们首先进行DNA提取和纯化,然后使用特定的方法对DNA进行增幅,然后进行测序反应,得到原始序列数据,最后通过生物信息学分析得到测序结果。
整个过程需要严格控制实验操作、仪器设备和实验环境,并在每个步骤中进行质控。
在本次测序中,我们采用了高通量测序技术,通过测序仪器的高效率和高准确性,成功得到了高质量的测序结果。
测序结果分析通过对测序结果的分析,我们获取了DNA序列的信息,包括序列长度、碱基组成、SNP等。
这些信息对于后续的研究和应用具有重要的意义。
我们可以利用这些序列信息进行功能基因预测、物种鉴定等研究,揭示生物体的遗传信息和进化过程。
反思测序结果可靠性尽管我们在测序过程中严格依照操作流程进行,但仍存在一定的误差和偏差。
首先,DNA提取和纯化过程中可能存在污染和损伤,导致测序结果的不准确性。
其次,测序反应中可能会出现PCR扩增的偏差或随机错误,进一步影响结果的精确性。
此外,测序仪器的读取速度和准确性也会对结果产生影响。
因此,我们需要在测序过程中不断优化和改进,提高测序结果的可靠性。
测序结果局限性测序结果的局限性主要体现在两个方面。
首先,测序技术限制了我们在一定时间内获取的序列长度,从而无法测序特别长的DNA片段。
其次,由于测序结果只是DNA序列的信息,我们无法直接获得DNA的三维结构和功能信息。
因此,在研究中需要结合其他技术手段和方法进行进一步分析和验证,以获取更全面和准确的结果。
结束语通过测序报告的总结与反思,我们对测序过程和结果有了更深入的认识。
我们深刻认识到测序的重要性和复杂性,对测序技术的优化和提高有了更高的要求。
希望在今后的研究中,能够进一步完善测序流程和技术,提高测序结果的可靠性和全面性,为科学研究和应用提供更准确和可靠的数据基础。

转录组测序报告原始数据预处理转录组测序是一种用于研究生物体特定时间点或条件下的基因表达的方法。
在进行转录组测序之前,需要对原始数据进行预处理,以确保数据的准确性和可靠性。
数据质量控制在转录组测序之前,应对原始数据进行质量控制。
常用的质量控制工具包括FastQC和Trimmomatic等。
通过这些工具,可以检查数据的质量,如测序错误率、测序深度、GC含量等,并根据需求进行相应处理,如剔除低质量的reads,修剪低质量的碱基等。
数据预处理在进行转录组测序分析之前,需要对原始数据进行一系列的预处理步骤。
常见的预处理步骤包括:1.去除适配体序列:适配体序列是在测序过程中引入的一段DNA序列,如果不去除适配体序列,会影响后续分析的准确性。
2.去除低质量的reads:低质量的reads指的是测序质量较差的序列,可能由于仪器误差或实验操作等原因造成。
通过设置阈值,可以将低质量的reads剔除,以提高数据的质量。
3.去除rRNA序列:在转录组测序中,rRNA序列往往占据了大部分的reads,如果不去除rRNA序列,会影响后续对其他RNA类别的分析。
可以使用软件工具,如Bowtie和SortMeRNA等,去除rRNA序列。
4.剔除PCR重复序列:PCR重复序列是由于PCR扩增过程导致的序列复制,会引入偏差。
通过标记和去除PCR重复序列,可以减少这种偏差。
5.序列长度过滤:根据实验需求和研究题目的特点,设置合适的序列长度过滤阈值,过滤掉过短或过长的reads。
6.序列比对:将清洗后的reads与参考基因组或转录组进行比对,以获取每个reads在基因组或转录组中的定位信息。
表达量分析转录组测序可以用来研究细胞中的基因表达水平。
表达量分析是转录组测序中的重要环节之一,可以通过表达量分析来确定不同基因的相对表达水平以及在不同条件下的差异表达。
转录本定量在表达量分析中,首先需要进行转录本定量。
转录本定量是指对转录组测序得到的reads进行比对,并根据比对结果计算每个转录本在给定样本中的表达量。
10x单细胞转录组结题报告一、项目背景与目的随着生物技术的飞速发展,单细胞转录组学已成为研究细胞类型、功能及其调控机制的重要手段。
10x Genomics公司作为该领域的佼佼者,其单细胞转录组技术广受关注。
本项目旨在利用10x单细胞转录组技术对某生物样本进行全面深入的分析,揭示其细胞类型的多样性、基因表达特征及其调控网络,为后续研究提供数据支持和理论依据。
二、实验设计与方法1.样本来源与处理:本研究所用样本来源于某生物组织,经过分离、纯化后获得单细胞悬液。
为确保细胞活力和完整性,采用特定的培养基和条件进行培养。
2.单细胞捕获与文库构建:利用10x Genomics公司的Chromium系统对单细胞进行捕获,并进行cDNA文库的构建。
通过特异性引物和PCR扩增,将cDNA 片段连接至含有细胞标签和UMI的测序接头,构建成可用于下游分析的文库。
3.测序与数据分析:采用Illumina测序平台对文库进行测序,获得原始数据。
利用10x Genomics提供的Cell Ranger软件对数据进行质量控制、细胞标签和UMI的识别、基因表达量的定量等分析。
同时,结合其他生物信息学工具和数据库,对数据进行深入挖掘和解读。
三、实验结果与讨论1.数据质量控制:通过对测序数据的分析,我们发现数据质量良好,细胞标签和UMI的识别准确率较高,基因表达量的定量结果可靠。
这为后续分析提供了有力的保障。
2.细胞类型鉴定:通过对基因表达谱的聚类分析,我们鉴定出多种细胞类型,包括已知类型和未知类型。
这些细胞类型在形态、功能及基因表达特征上均有所不同,反映了该生物样本的细胞多样性。
3.基因表达特征与调控网络:针对不同细胞类型,我们进一步分析了其基因表达特征和调控网络。
发现某些关键基因在不同细胞类型中的表达差异显著,可能与细胞的特定功能相关。
同时,我们还构建了基因调控网络,揭示了不同细胞类型中基因之间的相互作用关系。
4.与已有研究的比较:通过与已有研究进行比较,我们发现本研究的结果在细胞类型鉴定、基因表达特征及调控网络等方面均具有一定的创新性和补充性。
二、生物信息分析服务结果报告2.1 原始数据统计Sample Name Organ Name Total Reads Total Reads Pairs(Paired End)Insert(bp)Read Length (bp)Total Base Pairs (bp) s8_atcgt sepal 5,531,224 2,765,612 18096 530,997,504s8_gtcat leaf 6,768,440 3,384,220 18096 649,770,240s2_atcgt sepal 13,073,446 6,536,723 18076 993,581,896s2_gtcat leaf 14,067,128 7,033,564 18076 1,069,101,728Barcode信息:大岩桐萼片 barcodeatcgt大岩桐叶子 barcodegtcat2.2分析数据统计根据测序产生的序列文件,过滤掉低质量的序列后进行统计Sample Name Organ Name Filtered Reads Filtered Reads Pairs Read Length (bp)Total Filtered Base Pairs (bp) s8_atcgt sepal 4,886,722 2,443,361 96 469,125,312s8_gtcat leaf 5,998,374 2,999,187 96 575,843,904s2_atcgt sepal 10,751,756 5,375,878 76 817,133,456s2_gtcat leaf 12,944,588 6,472,294 76 983,788,6882.3 Solexa数据5’到3’端质量评估如果3’端的碱基质量值比较低,在后面的分析中需要切除3’端低质量碱基后进行分析。
质量分布图说明:1、横坐标数字代表每条read中的碱基位置,如:1代表read中的第一个碱基,依次类推一直到76是 read的最后一个碱基。
一、实验背景随着高通量测序技术的快速发展,转录组测序技术在生物学研究中的应用越来越广泛。
本研究旨在通过转录组测序技术,对某物种某组织在特定生理状态下的基因表达情况进行分析,揭示该物种在特定生理状态下的基因调控机制。
二、实验材料与方法1. 实验材料(1)样品:某物种某组织在特定生理状态下的样品。
(2)测序平台:Illumina HiSeq 2500。
2. 实验方法(1)RNA提取:采用Trizol法提取样品总RNA。
(2)RNA质检:利用NanoDrop 2000和Agilent 2100生物分析仪对RNA进行质检。
(3)cDNA文库构建:采用SMART-seq2技术构建cDNA文库。
(4)测序:利用Illumina HiSeq 2500进行测序。
(5)数据预处理:对原始测序数据进行质量过滤、拼接、去除接头序列等处理。
(6)转录组组装:利用Trinity软件对转录组进行组装。
(7)基因注释:将组装得到的转录本与NCBI RefSeq数据库进行比对,进行基因注释。
(8)基因表达分析:利用DESeq2软件进行差异表达基因(DEG)的筛选和表达量差异分析。
三、实验结果1. 转录组组装通过对测序数据进行组装,共得到12,345个转录本,其中长度大于200nt的转录本有11,275个。
2. 基因注释对组装得到的转录本进行基因注释,共注释到9,987个基因,其中6,872个基因有注释信息。
3. 差异表达基因筛选在特定生理状态下,共筛选出1,568个差异表达基因(DEG),其中上调基因935个,下调基因633个。
4. 差异表达基因功能富集分析对DEG进行GO(基因本体)和KEGG(京都基因与基因组百科全书)富集分析,发现DEG主要参与代谢、信号转导、细胞过程等生物学过程。
5. 差异表达基因共表达网络分析对DEG进行共表达网络分析,发现部分DEG在特定生理状态下具有协同调控作用。
四、结论本研究通过对某物种某组织在特定生理状态下的转录组测序和差异表达基因分析,揭示了该物种在特定生理状态下的基因调控机制。
转录组生物信息分析结题报告一、建库测序流程1. Total RNA样品检测2. 文库构建3. 库检4. 上机测序二、生物信息分析流程三、项目结果说明1. 原始序列数据2. 测序数据质量评估3. 参考序列比对分析4. 可变剪切分析5. 新转录本预测6. SNP和InDel分析7. 基因表达水平分析8. RNA-seq整体质量评估9. 基因差异表达分析10.差异基因GO富集分析11.差异基因KEGG富集分析12.差异基因蛋白互作网络分析13.DEU分析四、参考文献五、附录1. 文件目录列表2. 软件列表3. Methods英文版4. 结题报告PDF版北京诺禾致源生物信息科技有限公司一、建库测序流程从RNA样品到最终数据获得,样品检测、建库、测序每一个环节都会对数据质量和数量产生影响,而数据质量又会直接影响后续信息分析的结果。
因此,获得高质量数据是保证生物信息分析正确、全面、可信的前提。
为了从源头上保证测序数据的准确性、可靠性,诺禾致源对样品检测、建库、测序每一个生产步骤都严格把控,从根本上确保了高质量数据的产出。
流程图如下:1 Total RNA样品检测诺禾致源对RNA样品的检测主要包括4种方法:(1) 琼脂糖凝胶电泳分析RNA降解程度以及是否有污染(2) Nanodrop检测RNA的纯度(OD260/280比值)(3) Qubit对RNA浓度进行精确定量(4) Agilent 2100精确检测RNA的完整性2 文库构建样品检测合格后,用带有Oligo(dT)的磁珠富集真核生物mRNA(若为原核生物,则通过试剂盒去除rRNA来富集mRNA)。
随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs和DNA polymerase I合成二链cDNA,随后利用AMPure XP beads纯化双链cDNA。
转录组测序结题报告1.mRNA纯化:抽提得到的总RNA首先利用10U的DNaseI(Ambion,美国)在37℃消化1小时;然后利用Micropoly(A)PuristTM mRNA purification kit(Ambion,美国),进行mRNA纯化:把RNA稀释到250μl的体积,按照Kit的操作步骤(Cat.No:1919)进行;最后得到的mRNA用100μl预热的THE缓冲液洗脱,利用NanoDrop 进行定量。
2.cDNA合成:cDNA合成是在Ng等2005年发表的方法基础上改进而成(文献1,图1)。
第一链cDNA合成利用GsuI-oligo dT作为反转录引物,10μg的mRNA作为模板,用1000 单位的Superscript II reverse transcriptase (Invitrogen,美国)在42℃作用1小时完成;随后利用NaIO4(Sigma,美国)氧化mRNA的5’帽子结构,并连接生物素;通过Dynal M280磁珠(Invitrogen,美国)筛选连接了生物素的mRNA/cDNA,并通过碱裂解释放第一链cDNA;然后通过DNA ligase(TaKaRa,日本)在第一链cDNA的5’末端加上接头,然后通过Ex Taq polymerase (TaKaRa,日本)合成第二链cDNA。
最后通过GsuI酶切去除polyA和5’端接头。
图1. 全长cDNA合成示意图3.cDNA测序:合成的cDNA利用超声仪(Fisher)打断到300-500bp的范围,利用Ampure beads(Agencourt,美国)进行纯化。
随后纯化的cDNA利用TruSeq TM DNA XXmple Prep Kit – Set A (illumina,美国)制备文库,并利用TruSeq PE Cluster Kit (illumina,美国)进行扩增。
最后在illumina机器上进行测序反应。
转录组测序结题报告怎么看?点进来就知道啦!转录组测序(RNA-Seq)作为研究基因表达的利器,是发掘基因功能的重要途径。
随着RNA-Seq技术的普及,那么问题来了,很多不了解RNA-Seq的小伙伴,在点开结题报告的一瞬间,是不是满脑子的问号,不知所措呢?没关系!我们懂你!不了解RNA-Seq?不会看结题报告?莫慌,我们来给大家理头绪、划重点!首先,可将整个结题报告分成四个主要模块。
图 1 转录组测序结题报告主要模块差异基因的鉴定与功能富集分析是构成转录组文章的主体,数据挖掘与分析也是基于这两个模块进行,是结题报告的重心。
接下来详细告诉大家每个模块需要关注的重点内容。
原始数据整理与质量评估数据量的大小与测序质量的好坏是评判测序数据可靠性的重要标准。
▶数据量一般用Bases或Raw data表示,对于绝大部分物种来说,转录组测序6G数据量即可,若想获得更多低丰度基因的信息,可适当增加测序数据量。
▶数据质量主要包括碱基质量与碱基含量。
Illumina官方的碱基质量评价标准一般为Q30(即碱基错误识别率为0.1%),Q30的值越大越好,一般不能低于80%。
碱基含量即ATGC四种碱基所占的比例,除了前几个碱基位置之外,4种碱基的含量线条应平行且接近。
图 2 测序质量评估差异基因表达鉴定看基因的表达量与鉴定差异基因是做转录组测序的主要目的,生物学重复之间的相关性高低与差异基因鉴定的准确性息息相关。
▶样品相关性检验一般以矩阵图与PCA分析图展示。
在矩阵图中基因表达相近的样品会被聚到一起,生物学重复间相关系数越高越好,低于0.8表示相关性较差。
PCA分析图更加直观,可以把基因表达相关性好的样品展示到一起。
图3 样品相关性检验▶差异表达基因的鉴定在这里可以看到各个处理组与对照组之间基因的上、下调表达的信息。
从中查找所关注基因的表达情况。
显著差异基因判定标准:|log2 Foldchange|>1;P value < 0.05。
转录组测序结题报告1.mRNA纯化:抽提得到的总RNA首先利用10U的DNaseI(Ambion,美国)在37℃消化1小时;然后利用Micropoly(A)PuristTM mRNA purification kit(Ambion,美国),进行mRNA纯化:把RNA稀释到250μl的体积,按照Kit的操作步骤(Cat.No:1919)进行;最后得到的mRNA用100μl预热的THE缓冲液洗脱,利用NanoDrop 进行定量。
2.cDNA合成:cDNA合成是在Ng等2005年发表的方法基础上改进而成(文献1,图1)。
第一链cDNA合成利用GsuI-oligo dT作为反转录引物,10μg的mRNA作为模板,用1000 单位的Superscript II reverse transcriptase (Invitrogen,美国)在42℃作用1小时完成;随后利用NaIO4(Sigma,美国)氧化mRNA的5’帽子结构,并连接生物素;通过Dynal M280磁珠(Invitrogen,美国)筛选连接了生物素的mRNA/cDNA,并通过碱裂解释放第一链cDNA;然后通过DNA ligase(TaKaRa,日本)在第一链cDNA的5’末端加上接头,然后通过Ex Taq polymerase (TaKaRa,日本)合成第二链cDNA。
最后通过GsuI酶切去除polyA和5’端接头。
图1. 全长cDNA合成示意图3.cDNA测序:合成的cDNA利用超声仪(Fisher)打断到300-500bp的范围,利用Ampure beads(Agencourt,美国)进行纯化。
随后纯化的cDNA利用TruSeq TM DNA XXmple Prep Kit – Set A (illumina,美国)制备文库,并利用TruSeq PE Cluster Kit (illumina,美国)进行扩增。
最后在illumina机器上进行测序反应。
测序得到的数据统计见表1.表1. Solexa测序统计样品对照 1 2Reads数目(对) 5,500,000 10,254,848 11,160,428Clean data 5,442,815(98.96%)10,160,130(99.08%)10,998,951(98.55%)平均长度100 100 1005.EST拼装:利用trinity进行拼装。
共得到45,308个EST cluster(contigs)。
具体拼装结果见表2和图2。
表2. 拼装统计样品XXContig数目45,308Contig平均长度698Contig长度范围201-16,169图2. Contigs长度分布(横坐标为基因长度分布,纵坐标为基因数量分布)6.数据分析:6.1 基因预测:采用EMBOSS工具包(参考文献2)中的’GetORF’对拼装得到的contigs进行基因预测,从不同contigs中找到蛋白编码序列。
6.2 基因注释:将预测得到的蛋白编码序列与GenBank的NR、GO、KEGG、KOG等数据库利用blastp进行比对,条件为E value<1e-5,选择匹配最好的一项作为注释信息。
详细结果见annotation.xls,由左至右分别为拼接软件产生的contig名称、基因功能注释、ORF起始与终止位点坐标、正反义链、氨基酸长度、KOG分类。
6.3 GO分析:GO分析利用GoPipe(参考文献3)进行,预测蛋白首先与Swiss-Prot 和TrEMBL数据库进行比对,条件为blastp,E value<1e-5,然后比对结果利用GoPipe程序,根据gene2go,得到预测蛋白的GO信息。
共有4,823个预测蛋白,匹配28,168项GO terms,如图3所示。
详细结果见annotation.xls中“GO”sheet栏。
图3. GO分布6.4 代谢通路构建:利用KEGG数据库(参考文献3),将预测蛋白与KEGG数据库进行比对,条件为双向blast,E value <1e-3;得到预测蛋白的KO number,再根据KO number,获得预测蛋白参与的代谢通路信息。
结果共有2,706个蛋白获得了KO number,它们参与的代谢通路如如图4所示。
详细结果见annotation.xls中“KEGG pathway ”sheet栏。
图4. 编码蛋白所参与的代谢通路类别6.5 表达丰度分析:首先去除低值序列得到clean reads(图5),然后mapping 到拼接的contig上(图6,图7显示mapping的结果),统计每个conig中分别来自2个样品的reads数目,接着转换成RPKM(参考文献4),最后利用DEGseq 程序包中的MARS (MA-plot-based method with Random XXmpling model)模型(参考文献5),计算每个contig代表的基因在2个样品中的表达丰度差异,FDR 值小于0.001的即被认定为具有显著性差异。
详细结果见annotation.xls中“DGE”sheet栏或”express.xlsx”。
图5. 序列质量分析(clean reads为不含N且质量大于5的碱基数至少占全长的一半)图6. 测序饱和度分析(横坐标为reads number,纵坐标为gene number)图7.基因覆盖率统计样本间差异统计详见annotation.xls中“DGE”sheet栏:由左至右分别是基因名称、基因长度、样品A 统计reads数、样品A RPKM值、样品B统计reads 数、样品B RPKM 值、样品A相对样品B表达差异倍数(取Log值)、q-value、显著性判断。
表3. 样品间显著性差差异基因统计样品上调基因数(p<0.001)下调基因数(p<0.001)1/对照2,961 1,0052/对照2,257 362/1 3,352 2,541图8.上下调基因变化(横坐标为gene,纵坐标为统计值)6.6 富集分析:对于每一个代谢通路和GO类别,我们利用超几何分布统计,计算具有显著性表达差异的基因相对全部基因的显著富集情况。
结果在2个代谢通路和7个GO terms 中差异基因具有明显的富集(FDR<0.01) 详细结果见chayi-GO.xlsx 或者chayi-KEGG.xlsx表4. GO term 富集分析结果代谢通路P value 1/对照Carbohydrate Metabolism 0.003961 2/对照Translation1.02E-10 Cell Communication9.12E-06 Signaling Molecules and Interaction 0.002699 Cardiovascular Diseases 0.002838 Immune System 0.002838 2/1Translation0.000185 Energy Metabolism0.000377表5.代谢通路富集分析结果GO TermP value1/对照cell0.028283 metabolism0.028283 2/对照structural molecule activity 2.88E-34 biosynthesis 2.66E-11 cell0.000863 motor activity0.031182 2/1structural molecule activity 1.87E-33 biosynthesis 1.30E-28 cell0.000868 electron transport 0.01974 metabolism0.0313336.7 客户定制分析:6.7.1 调控途径构建于分析Carotenoid biosynthesis 代谢途径相关基因的富集整理。
表6 Carotenoid biosynthesis 代谢途径整理6.7.2 SSR分子标记筛选详细结果见SSR.xlsx6.7.3 SNP鉴定与筛选详细结果见SNP.xlsx7. FTP文件说明所有分析结果都在FTP的对应文件夹中,具体的解释详见“RNA-Seq相关说明”。
8.参考文献:1. Rice, P., I. Longden, and A. Bleasby, EMBOSS: the European Molecular Biology OpenSoftware Suite. Trends Genet, 2000. 16(6): p. 276-7.2. Chen, Z.-Z.X., C.-H. Zhu, S., GoPipe: streamlined gene ontology annotation for batchanonymous sequences with statistics. PROGRESS IN BIOCHEMISTRY AND BIOPHYSICS, 2005. 32(2): p. 187-190.3. KanehiXX, M., et al., KEGG for representation and analysis of molecular networks involvingdiseases and drugs. Nucleic Acids Res. 38(Database issue): p. D355-60.4. Mortazavi, A., et al., Mapping and quantifying mammalian transcriptomes by RNA-Seq. NatMethods, 2008. 5(7): p. 621-8.5. Wang, L., et al., DEGseq: an R package for identifying differentially expressed genes fromRNA-seq data. Bioinformatics. 26(1): p. 136-8.。