诺禾致源真核无参转录组生物信息分析结题报告2013年8月
- 格式:pdf
- 大小:13.19 MB
- 文档页数:38

诺禾致源LncRNA信息分析内容LncRNA分析内容注:以下标红需要多样本数据支持一、标准信息分析内容:1.测序数据质量评估2.参考序列比对分析3.基因表达水平分析4.RNA--‐seq整体质量评估5.序列拼接组装(cufflinks + S cripture两种拼接方法 )6.LncRNA位点筛选(可选intergenic lncRNA, intronic LncRNA, antisense lncRNA分别计算费用)6.1长度筛选(>=200bp)6.2Exon个数筛选(>=2)6.3Reads覆盖度筛选(>=3)6.4位置筛选(对不同种类的lncRNA采用不同的位置筛选条件)6.5编码潜能筛选(四种方法取交集:pfam蛋白结构域筛选、CPC 筛选、CNCI筛选、PhyloCSF)7. 筛选得到的LncRNA描述性统计分析7.1 L ncRNA长度分布7.2 E xon个数分布7.3 已知和新预测lncRNA分类8. LncRNA保守性分析8.1 位点保守性分析8.2 序列保守性分析9. LncRNA表达水平分析9.1 差异表达水平分析*(>=2个样本)9.2 组织或表型特异性表达分析*(>=3个组织或者表型)10.LncRNA靶基因预测10.1 C is作用靶基因10.2 T rans作用靶基因*(>=2个样本)11. L ncRNA靶基因功能注释11.1 C is作用功能注释11.2 T rans作用功能注释*(>=5个样本)12. 差异表达或特异性表达的靶基因功能分析*(>=2个样本)12.1 G O富集分析12.2 K EGG富集分析12.3差异LincRNA与靶基因调控网络分析二、个性化分析内容:1. l ncRNA g ene与转录组coding g ene比较分析(需要转录组数据的分析结果)1.1 长度分布比较1.2 表达水平比较1.3 组织或表型特异性比较1.4 保守性比较2. L ncRNA浏览器辅助建立。

诺禾致源再添一作成果组装新策略完成犬蝠基因组精细图谱并揭示食果蝙蝠免疫基因演化新机制
近些年来,随着基因组学技术的发展和普及,动物的基因组结构如犬、蝠等动物的基因组分析也开始受到越来越多关注。
基于诺禾致源和其他研
究机构相关研究成果,我们最近又实施了一次新的策略来完成犬蝠基因组
精细图谱,并发现了食果蝙蝠免疫基因演化的新机制。
首先,为了完成精细图谱,我们首先采用了诺禾致源的高通量测序技术,对52份犬蝠基因组和17份食果蝙蝠基因组进行测序与建库,利用该
技术最终获得了更为精细的基因组图谱。
同时,利用该技术也可以获得更
准确的免疫基因定位信息,从而揭示了食果蝙蝠抗病毒免疫基因演化机制。
其次,我们还采用了高通量比对技术来获得犬蝠和食果蝙蝠的基因表
达水平。
我们在犬蝠和食果蝙蝠的基因组中从各种环境中提取和准备组织
样本,然后用高通量比对技术检测各个基因的表达水平,从而更准确地揭
示基因演化的机制。
最后,我们还采用了分子生物学技术,用单链接聚合酶链反应法(SLiP-PCR)来分析食果蝙蝠的基因组片段,获得更多的基因组信息。
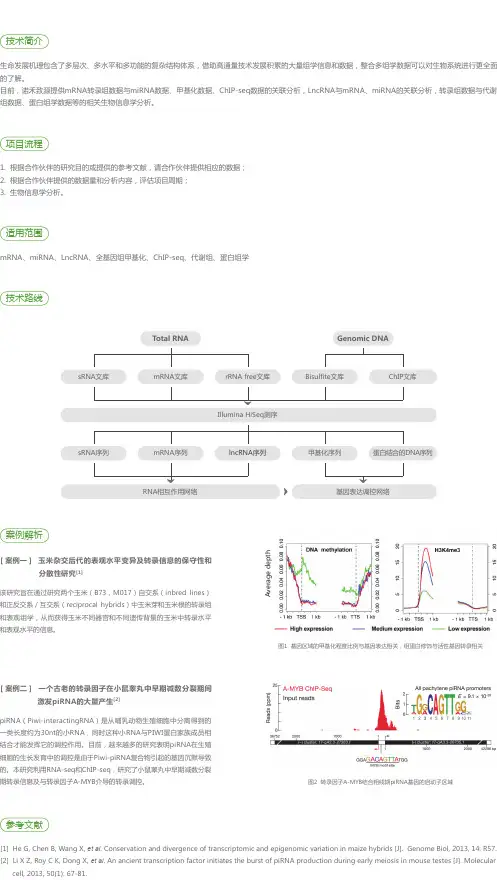
Total RNA Genomic DNAsRNA文库sRNA序列Bisulfit e文库甲基化序列mRNA文库mRNA序列Illumina HiSeq测序ChIP文库蛋白结合的DNA序列rRNA fr ee文库lncRNA序列生命发展机理包含了多层次、多水平和多功能的复杂结构体系,借助高通量技术发展积累的大量组学信息和数据,整合多组学数据可以对生物系统进行更全面的了解。
目前,诺禾致源提供mRNA转录组数据与miRNA数据、甲基化数据、ChIP-seq数据的关联分析,LncRNA与mRNA、miRNA的关联分析,转录组数据与代谢组数据、蛋白组学数据等的相关生物信息学分析。
1. 根据合作伙伴的研究目的或提供的参考文献,请合作伙伴提供相应的数据;2. 根据合作伙伴提供的数据量和分析内容,评估项目周期;3. 生物信息学分析。
项目流程mRNA、miRNA、LncRNA、全基因组甲基化、ChIP-seq、代谢组、蛋白组学适用范围技术路线技术简介参考文献[1] He G, Chen B, Wang X, et al . Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids [J]. Genome Biol, 2013, 14: R57.[2] Li X Z, Roy C K, Dong X, et al . An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes [J]. Molecular cell, 2013, 50(1): 67-81.案例解析[案例一] 玉米杂交后代的表观水平变异及转录信息的保守性和 分散性研究[1]该研究旨在通过研究两个玉米(B73,M017)自交系(inbred lines)和正反交系/互交系(reciprocal hybrids)中玉米芽和玉米根的转录组和表观组学,从而获得玉米不同器官和不同遗传背景的玉米中转录水平和表观水平的信息。

·815·水稻响应白叶枯病菌侵染的转录组分析张宇1,2,王金开2,陈小林1*,李其利1,唐利华1,黄穂萍1,郭堂勋1,玉延华2*(1广西农业科学院植物保护研究所/农业农村部华南果蔬绿色防控重点实验室/广西作物病虫害生物学重点实验室,广西南宁530007;2广西大学生命科学与技术学院,广西南宁530004)摘要:【目的】分析白叶枯病菌(Xanthomonas oryzae pv.oryae ,Xoo )侵染不同时间点的水稻转录组数据,了解其响应侵染过程中抗病相关基因的表达情况,为明确水稻对白叶枯病菌感病机制以及培育新的抗白叶枯病水稻品种提供理论参考。
【方法】以含有广谱抗性基因Xa23的水稻品系CBB23为试验材料,对其叶片接种高致病性的野生型Xoo 菌株N3-1,将接种0、48和72h 水稻样品总RNA 进行转录组测序(RNA-Seq )。
以|log 2Fold Change|≥1,且错误发现率(False discovery rate ,FDR )<0.01为标准筛选差异表达基因(Differentially expressed genes ,DEGs ),并进行GO 功能注释和KEGG 通路富集分析;通过实时荧光定量PCR (qRT-PCR )验证RNA-Seq 结果的可靠性。
【结果】RNA-Seq 转录组测序结果共获得约54.63Gb Clean data ,样品平均数据量约为6.07Gb ,Q30碱基准确率在94.34%及以上。
与对照组0h 相比,水稻CBB23在接种48和72h 两个时间点分别鉴定出2361和2117个DEGs ,其中48h 上调基因1650个、下调基因711个,72h 上调基因1440个、下调基因677个。
GO 富集结果表明,水稻的DEGs 显著富集于芳香族氨基酸的代谢及二萜类的生物合成。
KEGG 通路富集结果表明,DEGs 富集于苯丙氨酸和苯丙素类的生物合成。


陆地棉基因组测序揭示四倍体棉进化与纤维发育机制Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement研究对象:陆地棉遗传标准系TM-1期刊:Nature Biotechnology影响因子:41.514合作单位:南京农业大学发表时间:2015年4月摘 要Upland cotton is a model for polyploid crop domestication and transgenic improvement. Here we sequenced the allotetraploid Gossypium hirsutum L. acc. TM-1 genome by integrating whole-genome shotgun reads, bacterial artificial chromosome (BAC)-end sequences and genotype-by-sequencing genetic maps. We assembled and annotated 32,032 A-subgenome genes and 34,402 D-subgenome genes. Structural rearrangements, gene loss, disrupted genes and sequence divergence were more common in the A subgenome than in the D subgenome, suggesting asymmetric evolution. However, no genome-wide expression dominance was found between the subgenomes. Genomic signatures of selection and domestication are associated with positively selected genes (PSGs) for fiber improvement in the A subgenome and for stress tolerance in the D subgenome. This draft genome sequence provides a resource for engineering superior cotton lines.关键词陆地棉;de novo;四倍体研究背景陆地棉(Gossypium hirsutum L.)隶属锦葵目(Malvales),锦葵科(Malvaceae),棉属(Gossypium),因最早在美洲大陆种植而得名,是世界上最重要的棉花栽培品种,占全球棉花种植面积的90%以上。


图2 显著富集的KEGG通路图1 A 新合成异源多倍体小麦中12种差异表达基因 B 非加性表达miRNA与亲本显性表达miRNA的 等级聚类分析和两者的关联图3 菟丝子与拟南芥、番茄转移RNA和非转移RNA的表达和富集分析. mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat [J]. The Plant Cell, 2014: tpc. 114.124388.. Effects of Tris(1,3-dichloro-2-propyl) Phosphate (TDCPP) in Tetrahymena Thermophila: [3] Kim G, LeBlanc M L, et al. Genomic-scale exchange of mRNA between a parasitic plant and its hosts [J]. Science, 2014, 345图1 人类着床前胚胎在发育过程中的基因表达聚类分析案例二 利用单细胞RNA-seq手段追踪人类和小鼠胚胎的遗传发育本研究采用单细胞RNA-seq技术对人类和小鼠胚胎从卵母细胞到桑椹胚阶段的转录组动态进行了全面的分析。
通过权重基因共表达网络分析(WGCNA),发现通过少量共表达基因功能性模块就可以简明地描绘每个发育阶段。
小鼠和人类植入前胚胎比较分析结果表明,人类和小鼠之间发育特异性和时序上存在差异,大多数的人类阶段特异性模块(9 个)在物种间具有保守性。
此外,鉴别出人类和小鼠网络中一些保守的模块成员:hub选基因可能参与调控哺乳动物着床期以前的胚胎发育过程。
图2 人类胚胎基因共表达模块[1] Yan L, Yang M, Guo H, et al. Single-cell RNA-Seq profling of human preimplantation embryos and embryonic stem cells [J]. Nature structural & molecular biology, 2013.[2] Xue Z, Huang K, Cai C, et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing [J]. Nature, 2013, 500(7464): 593-597.图1 栽培番茄和野生番茄构建系统进化树案例二 比较转录组揭示野生种苎麻向栽培种苎麻进化机制诺禾致源携手中国农业科学院麻类研究所,利用比较转录组解析栽培种苎麻和野生种苎麻在基因序列水平上的自然变异,找到13个受到正向选择的基因,这些差异可能与植物的抗病或者抗逆境有关,推测生物和非生物胁迫在苎麻的驯化过程中可能起着非常重要的作用;另外,两个受正向选择的基因与苎麻的纤维产量相关,主要是人工选择导致的。

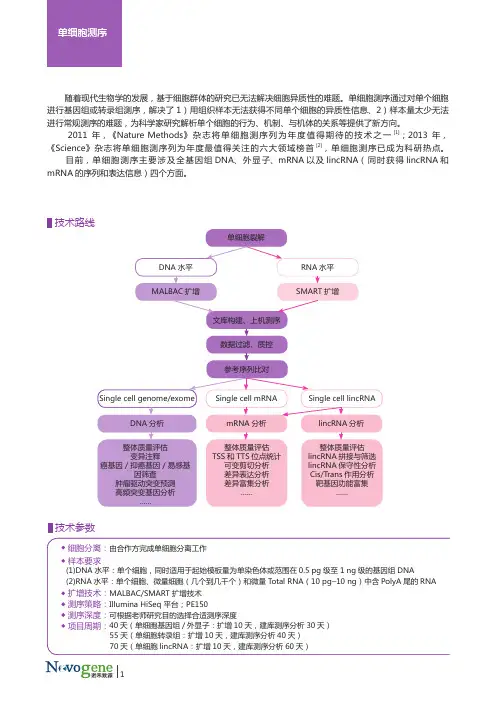
随着现代生物学的发展,基于细胞群体的研究已无法解决细胞异质性的难题。
单细胞测序通过对单个细胞进行基因组或转录组测序,解决了1)用组织样本无法获得不同单个细胞的异质性信息、2)样本量太少无法进行常规测序的难题,为科学家研究解析单个细胞的行为、机制、与机体的关系等提供了新方向。
2011 年,《Nature Methods》杂志将单细胞测序列为年度值得期待的技术之一[1];2013 年,《Science》杂志将单细胞测序列为年度最值得关注的六大领域榜首[2],单细胞测序已成为科研热点。
目前,单细胞测序主要涉及全基因组DNA、外显子、mRNA以及lincRNA(同时获得lincRNA和mRNA的序列和表达信息)四个方面。
技术路线技术参数产品优势最严格的数据指控标准诺禾建立了新一代高通量测序平台,包括10台Hiseq X 测序仪,10台Hiseq 2500,从样品检测到文库构建采用最严格的指控标准,从源头保证了数据的可靠性。
采用国际认可的全基因组扩增技术DNA 水平:诺禾致源采用多次退火环状循环扩增技术(MALBAC),MALBAC 扩增具有低偏倚性。
最新《Science》文章比较MALBAC 与MDA 扩增技术,发现MALBAC 扩增后的数据与混合建库的数据质量更为接近(如图1)[3]。
该技术应用于单精子测序,研究成果[4]已在《Science》杂志发表。
RNA 水平:诺禾致源采用SMARTer 扩增技术(美国Clontech 公司原装试剂盒),能够对单个细胞、微量细胞(几个到几千个)和微量Total RNA(10 pg-10 ng)中含PolyA 尾的RNA 全长进行准确而高效地扩增,扩增后的cDNA 片段适用于Illumina 建库测序平台,为单细胞样本的RNA-seq 分析提供了利器。
图2 SMARTer 扩增无3'和5'端偏好性定制化信息分析针对不同项目,除了进行标准信息分析外,通过和合作伙伴共同探讨,制定可行的个性化信息分析方案,整合各种主流软件,优化分析结果,以确保结果的准确性及创新性。
诺禾致源rna测序方法什么是诺禾致源RNA测序方法?诺禾致源(Nugen)是一家生物技术公司,专注于开发和提供先进的分子生物学工具和服务。
他们的RNA测序方法被称为诺禾致源RNA测序方法,它是一种能够高效、准确地分析RNA样本的技术。
RNA测序是一种重要的分子生物学工具,用于确定细胞中存在的RNA分子的类型和数量。
通过诺禾致源RNA测序方法,可以在高通量、高灵敏度和高特异性的条件下,对RNA样本进行全面的测序分析。
诺禾致源RNA测序方法的一大特点是其使用了单分子扩增技术。
这种方法能够从单个RNA分子开始扩增,避免了传统方法中的批量扩增带来的偏差和误差。
此外,诺禾致源RNA测序方法还采用了特殊的试剂和引物,使测序的覆盖度、准确性和重复性得到了很大的提高。
使用诺禾致源RNA测序方法进行RNA测序需要经过以下几个主要步骤:1. 样本准备:首先,需要从目标细胞或组织中提取RNA样本。
样本准备的质量和纯度对后续的测序分析非常重要。
2. RNA逆转录:接下来,将提取到的RNA样本逆转录为cDNA。
这涉及到使用逆转录酶和引物,将RNA转录为cDNA,以便后续的扩增和测序。
3. DNA扩增:在RNA逆转录的基础上,使用诺禾致源的单分子扩增技术,将cDNA进行扩增。
通过这种方法,每个cDNA分子都能够独立地进行扩增,从而避免了传统批量扩增中的偏差和误差。
4. 测序准备:将扩增后的DNA片段进行准备,包括对DNA进行片段化、链接适配体和固定到测序芯片上。
这些步骤能够为后续的测序提供必要的样品准备。
5. 测序分析:最后,通过高通量测序技术对固定在芯片上的DNA片段进行测序。
通过诺禾致源的RNA测序方法,可以获得高质量、高覆盖度的测序数据。
除了这些基本步骤外,诺禾致源RNA测序方法还可以与其他分析技术和工具相结合,以实现更全面的RNA分析。
例如,可以使用生物信息学工具对测序数据进行处理和分析,以获得RNA的表达水平、剪接变异等信息。
江苏农业学报(Jiangsu J.of Agr.Sci.),2012,28(4):697 702常闪闪,肖姗姗,张磊,等.水稻OsSsr1基因的生物信息学分析、亚细胞定位及其表达模式[J ].江苏农业学报,2012,28(4):697-702.水稻OsSsr1基因的生物信息学分析、亚细胞定位及其表达模式常闪闪,肖姗姗,张磊,李文奇,刘凤权,邵敏(南京农业大学植物保护学院,农作物生物灾害综合治理教育部重点实验室,江苏南京210095)收稿日期:2012-05-03基金项目:国家“863”项目(2008AA10Z108、2007AA10Z188);国家转基因生物新品种培育项目(2009ZX08001-005B )作者简介:常闪闪(1986-),女,河南许昌人,硕士研究生,研究方向为分子和生理植物病理学。
(E-mail )2009102043@njau.edu.cn通讯作者:邵敏,(E-mail )minshao@njau.edu.cn 摘要:为了探明盐敏感相关基因[OsSsr1(Oryza sativa L.salt -sensitive related gene 1,GenBank 登录号为NM_001065574)]的亚细胞定位及在水稻不同组织和各种逆境胁迫下的表达模式,通过生物信息学手段、洋葱表皮亚细胞定位、RiceXPro 数据库和Real-time PCR 对其进行研究。
序列分析表明OsSsr1的开放阅读框为2004bp ,编码一个由667个氨基酸组成并含有5个跨膜区的蛋白质,该蛋白质N 端含有一个信号肽;其启动子序列包含TGACG-motif 、ABRE 、TCA-element 和W box 等多个与逆境相关的顺式作用元件。
GFP 融合表达显示,OsSSR1蛋白质定位于细胞膜。
RiceXPro 数据库中数据分析表明,OsSsr1基因在水稻不同发育期的不同组织中均有表达,在叶中的表达量最高,胚中的表达量最低。
首页 科技服务 测序指南 基因课堂 市场活动与进展 文章成果 关于我们全基因组选择1. Meuwissen T H, Hayes B J, Goddard M E.Prediction of total genetic value using genome-wide dense marker maps[J]. Genetics, 2001, 157(4): 1819 1829. 阅读原文>>2. Haberland A M, Pimentel E C G, Ytournel F, et al. Interplay between heritability, genetic correlation and economic weighting in a selection index with and without genomic information[J]. Journal of Animal Breeding and Genetics, 2013, 130(6): 456-467. 阅读原文>>3. Wu X, Lund M S, Sun D, et al. Impact of relationships between test and training animals and among training animals on reliability of genomic prediction[J]. Journal of Animal Breeding and Genetics, 2015, 132(5): 366-375. 阅读原文>>4. Goddard M E ,Hayes BJ. Genomic selection [J]. Journal of Animal Breeding and Genetics,2007,124:323:330. 阅读原文>>5. Heffner E L, Sorrells M E, Jannink J L. Genomic selection for crop improvement [J]. Crop Science, 2009, 49(1): 1-12. 阅读原文>>参考文献全基因组选择简介Meuwissen等[1]在2001年首次提出了基因组选择理论(Genomic selection , GS),即利用具有表型和基因型的个体来预测只具有基因型不具有表型值动植物的基因组育种值(GEBV)。
剑指NG!诺禾致源助力鉴定影响调控元件的人类SNPs国庆假期,小编在翻朋友圈的时候,看到远在荷兰留学的师兄也晒了自己的出游美照,花海风车大牧场真是一个都不少。
听师兄说,荷兰不光景色好,生活节奏慢,科研水平还挺高,动不动就能发个CNS 什么的。
这不,今年7月份,荷兰癌症研究所 Bas van Steensel 教授研究团队,借助诺禾强大的测序平台和专业的信息分析,成功开发出一种基于高通量测序的方法来鉴定影响转录调控元件活性的 SNPs,其研究成果发表在国际顶级期刊 Nature Genetics[1]!下面就让小编带大家一起学习一下吧~研究背景传统的 GWAS 分析所得到的 SNPs 大多处于非编码区,通常只能说明与疾病相关的突变“在哪儿”,但是难以完全解释这些突变“有什么用”。
越来越多的研究表明,非编码区的突变通过影响基因的表达调控从而导致疾病和表型的多样性[2~4]。
而基于报告实验的功能性筛选其通量很小,难以测试大量的SNPs 对调控元件活性的可能影响。
为此,作者开发了一种调控元件调查(survey of regulatory elements, SuRE)报告技术,以高通量测序的方法,对590万个SNPs 进行了鉴定,并从中筛选出对启动子和增强子活性具有调控作用的 SNPs,解决了这一难题。
研究方法▪样本选择:使用HG02601、GM18983、HG01241、HG03464 四个样本进行基因组 DNA 的获取。
▪文库构建及分析思路:将基因组DNA 打断为大约300bp 的片段来构建SuRE 文库。
这些片段与不含启动子,并携带了不同的barcode 的质粒进行连接。
通过基因组测序的方法,将 barcode 信息与片段上的SNP 信息进行标定。
将构建好的质粒转染到K562 及HepG2 细胞系中,并对细胞进行转录组测序。
根据barcode 表达量差异,寻找直接调控启动子/增强子活性的SNP 位点,并将其定义为raQTLs。
真核无参转录组生物信息分析结题报告建库测序流程Total RNA样品检测文库构建上机测序北京诺禾致源生物信息科技有限公司一、建库测序流程从RNA样品到最终数据获得,样品检测、建库、测序每一个环节都会对数据质量和数量产生影响,而数据质量又会直接影响后续信息分析的结果。
为了从源头上保证测序数据的准确性、可靠性,诺禾致源对样品检测、建库、测序每一个生产步骤都严格把控,从根本上确保了高质量数据的产出。
实验流程图如下:1 Total RNA样品检测诺禾致源对RNA样品的检测主要包括4种方法:(1) 琼脂糖凝胶电泳分析RNA降解程度以及是否有污染(2) Nanodrop检测RNA的纯度(OD260/280比值)(3) Qubit对RNA浓度进行精确定量(4) Agilent 2100精确检测RNA的完整性2 文库构建及库检样品检测合格后,用带有Oligo(dT)的磁珠富集真核生物mRNA(若为原核生物,则通过试剂盒去除rRNA来富集mRNA)。
随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs、RNase H和DNA polymerase I合成二链cDNA,随后利用AMPure XP beads纯化双链cDNA。
纯化的双链cDNA再进行末端修复、加A尾并连接测序接头,然后用AMPure XP beads进行片段大小选择,最后进行PCR富集得到最终的cDNA文库。
文库构建完成后,先使用Qubit2.0进行初步定量,稀释文库至1ng/ul,随后使用Agilent 2100对文库的insert size进行检测,insert size符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2nM),以保证文库质量。
文库构建原理图如下:3 上机测序库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina HiSeq/MiSeq测序。
真核无参转录组生物信息分析结题报告建库测序流程Total RNA样品检测文库构建上机测序北京诺禾致源生物信息科技有限公司一、建库测序流程从RNA样品到最终数据获得,样品检测、建库、测序每一个环节都会对数据质量和数量产生影响,而数据质量又会直接影响后续信息分析的结果。
为了从源头上保证测序数据的准确性、可靠性,诺禾致源对样品检测、建库、测序每一个生产步骤都严格把控,从根本上确保了高质量数据的产出。
实验流程图如下:1 Total RNA样品检测诺禾致源对RNA样品的检测主要包括4种方法:(1) 琼脂糖凝胶电泳分析RNA降解程度以及是否有污染(2) Nanodrop检测RNA的纯度(OD260/280比值)(3) Qubit对RNA浓度进行精确定量(4) Agilent 2100精确检测RNA的完整性2 文库构建及库检样品检测合格后,用带有Oligo(dT)的磁珠富集真核生物mRNA(若为原核生物,则通过试剂盒去除rRNA来富集mRNA)。
随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs、RNase H和DNA polymerase I合成二链cDNA,随后利用AMPure XP beads纯化双链cDNA。
纯化的双链cDNA再进行末端修复、加A尾并连接测序接头,然后用AMPure XP beads进行片段大小选择,最后进行PCR富集得到最终的cDNA文库。
文库构建完成后,先使用Qubit2.0进行初步定量,稀释文库至1ng/ul,随后使用Agilent 2100对文库的insert size进行检测,insert size符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2nM),以保证文库质量。
文库构建原理图如下:3 上机测序库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina HiSeq/MiSeq测序。
北京诺禾致源生物信息科技有限公司二、生物信息分析流程对于无参考基因组的转录组分析,可先将测序所得的序列拼接成转录本,以转录本为参考序列,进行后续分析。
信息分析流程图如下:三、结果展示及说明1 原始序列数据高通量测序(如Illumina HiSeq TM2000/Miseq TM)得到的原始图像数据文件经CASAVA碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为 Raw Data或Raw Reads,结果以 FASTQ (简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
FASTQ格式文件中每个read由四行描述,如下:@H W I-S T1276:71:C1162A C X X:1:1101:1208:24581:N:0:C G A T G TN A A G A A C A C G T T C G G T C A C C T C A G C A C A C T T G T G A A T G T C A T G G G A T C C A T+#55???B B B B B?B A@D E E F F C F F H H F F C F F H H H H H H H F A E0E C F F D/A E H H其中第一行以“@”开头,随后为Illumina 测序标识别符(Sequence Identifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为Illumina 测序标识别符(选择性部分);第四行是对应碱基的测序质量,该行中每个字符对应的 ASCII 值减去 33,即为对应第二行碱基的测序质量值。
Illumina测序标识符详细信息如下:HWI-ST1276Instrument – unique identifier of the sequencer71run number – Run number on instrumentC1162ACXX FlowCell ID – ID of flowcell1LaneNumber – positive integer1101TileNumber – positive integer1208X – x coordinate of the spot. Integer which can be negative2458Y – y coordinate of the spot. Integer which can be negative1ReadNumber - 1 for single reads; 1 or 2 for paired endsN whether it is filtered - NB:Y if the read is filtered out, not in the delivered fastq file, N otherwise0control number - 0 when none of the control bits are on, otherwise it is an even number CGATGThao Illumina index sequences2 测序数据质量评估2.1 测序错误率分布检查如果测序错误率用e表示,Illunima HiSeq TM2000/Miseq TM的碱基质量值用Q phred表示,则有:Q phred=-10log10(e)。
Illunima Casava 1.8版本碱基识别与Phred分值之间的简明对应关系见下表:Phred分值不正确的碱基识别碱基正确识别率Q-sorce 101/1090%Q10201/10099%Q20301/100099.9%Q30401/1000099.99%Q40对于RNA-seq技术,测序错误率分布具有两个特点,具体见图1:(1)测序错误率会随着测序序列(Sequenced Reads)的长度的增加而升高,这是由于测序过程中化学试剂的消耗而导致的,并且为illumina高通量测序平台都具有的特征(Erlich and Mitra, 2008; Jiang et al.)。
(2)前6个碱基的位置也会发生较高的测序错误率,而这个长度也正好等于在RNA-seq建库过程中反转录所需要的随机引物的长度。
所以推测这部分碱基的测序错误率较高的原因为随机引物和RNA模版的不完全结合所致(Jiang et al.)。
一般情况下,单个碱基位置的测序错误率应该低于1%。
图1 测序错误率分布图横坐标为reads的碱基位置,纵坐标为单碱基错误率前100bp 为双端测序序列的第一端测序 Reads的错误率分布情况,后100bp为另一端测序reads 的错误率分布情况。
2.2 A/T/G/C 含量分布检查GC 含量分布检查用于检测有无AT 、GC 分离现象。
对于RNA-seq 来说,因随机性打断及G/C 和A/T 含量分别相等的原则,理论上GC 及AT 含量每个测序循环上应分别近似相等(若为链特异性建库,可能会出现AT 分离和/或GC 分离),且整个测序过程基本稳定不变,呈水平线。
但在现有的高通量测序技术中,反转录成cDNA 时所用的6bp 的随机引物会引起前几个位置的核苷酸组成存在一定的偏好性,这种波动属于正常情况。
如图 2 所示:图2 GC 含量分布图横坐标为reads 的碱基位置,纵坐标为单碱基所占的比例;不同颜色代表不同的碱基类型前100bp 为双端测序序列的第一端测序Reads 的GC 分布情况,后100bp 为另一端测序reads 的GC 分布情况。
2.3 测序数据过滤测序得到的原始测序序列(Sequenced Reads)或者 raw reads,里面含有带接头的、低质量的reads,如图3所示。
为了保证信息分析质量,必须对raw reads过滤,得到clean reads,后续分析都基于 clean reads。
数据处理的步骤如下:(1) 去除带接头(adapter)的reads;(2) 去除N(N表示无法确定碱基信息)的比例大于10%的reads;(3) 去除低质量reads(质量值sQ <= 5的碱基数占整个read的50%以上的reads)。
RNA-seq的接头信息:TruSeq® RNA and DNA Sample Prep Kits (v1 and v2) 2,5TruSeq Universal Adapter(5’端接头)5’ AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTruSeq Adapter(3’端接头,带下划线的 6bp 的碱基为 Index,共有24种Index)5’ GATCGGAAGAGCACACGTCTGAACTCCAGTCACATCACGATCTCGTATGCCGTCTTCTGCTTG图3 原始数据组成不同颜色的比例分别代表不同成分比例(1)Adapter related:因有接头,过滤掉的 reads数及其占总 raw reads数的比例。
(2)Containing N:因 N含量超过 10%,过滤掉的 reads数及其占总raw reads数的比例。
(3)Low quality:因低质量,过滤掉的reads数及其占总raw reads数的比例。
(4)Clean reads:最终得到的 clean reads 数及其占总 raw reads 数的比例。
2.4 测序数据质量情况汇总样品测序产出数据质量评估情况详见表1。
表1 数据产出质量情况一览表Sample Raw Reads Clean reads Clean bases Error(%)Q20(%)Q30(%)GC(%) VIYCK_15145489450205417 5.02G0.0397.8992.4845.55 VIYCK_25145489450205417 5.02G0.0397.6892.7845.45 VIYCd_14283315141793838 4.18G0.0398.4694.4645.45 VIYCd_24283315141793838 4.18G0.0397.5492.9745.38 Sample: 样品名。
1为左端reads,2为右端reads。
样品的 clean reads 总数为左端+右端。
Raw reads:统计原始序列数据,以四行为一个单位,统计每个文件的测序序列的个数。
Clean reads:计算方法同 Raw Reads、Raw bases,只是统计的文件为过滤后的测序数据。