红外光谱分析峰值
- 格式:docx
- 大小:23.01 KB
- 文档页数:6

红外吸收光谱特征峰1. 水平振动峰:大部分物质在红外光谱中显示出实数振动峰,这些峰通常位于1500-4000 cm^-1区间。
在这个区间内,主要的振动模式有:C-H拉伸振动,C=O伸缩振动,C-N伸缩振动和O-H伸缩振动等。
2. 弯曲振动峰:这些峰通常位于500-1500 cm^-1区间,代表物质中相对较低能量的振动模式。
其中,主要的弯曲振动包括:C-H弯曲振动、O-H弯曲振动和C-N弯曲振动等。
3. 拉曼峰:拉曼光谱是一种与红外光谱类似的光谱,主要用于研究物质的分子振动。
拉曼光谱中的峰通常位于200-4000 cm^-1区间,包括了与红外光谱重叠的水平和弯曲振动。
4. 振动-转动峰:当分子既有振动运动又有转动运动时,红外光谱中会出现振动-转动峰。
这些峰通常位于0-500 cm^-1区间,具有特定的振动和转动组合频率,可以用来确定分子的对称性。
5. 过渡金属峰:一些过渡金属化合物在红外光谱中显示出独特的吸收峰。
这些峰通常位于400-2000 cm^-1区间,对应于金属-配体之间的振动模式。
6. 质子峰:质子(H+)在红外光谱中呈现为一个孤立线峰。
质子峰的位置通常在1500-2500 cm^-1之间,变化范围较大,取决于质子的环境。
红外吸收光谱中的这些特征峰可以提供物质的结构、键合和功能基团等信息。
通过分析化合物在红外光谱中的峰值位置和形状,可以确定其化学组成和化学结构,实现化合物的鉴定和分析。
同时,红外光谱还可以用于跟踪反应过程、监测化学变化和定量分析等方面。
这些特征峰在各个研究领域,如有机化学、材料科学和生物化学等中都有广泛的应用。
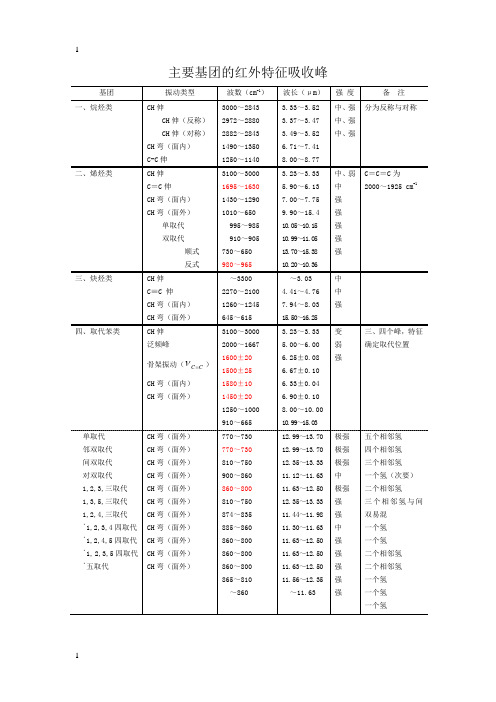
主要基团的红外特征吸收峰基团振动类型波数(cm-1)波长(μm)强度备注一、烷烃类CH伸CH伸(反称)CH伸(对称)CH弯(面内)C-C伸3000~28432972~28802882~28431490~13501250~11403.33~3.523.37~3.473.49~3.526.71~7.418.00~8.77中、强中、强中、强分为反称与对称二、烯烃类CH伸C=C伸CH弯(面内)CH弯(面外)单取代双取代顺式反式3100~30001695~16301430~12901010~650995~985910~905730~650980~9653.23~3.335.90~6.137.00~7.759.90~15.410.05~10.1510.99~11.0513.70~15.3810.20~10.36中、弱中强强强强强C=C=C为2000~1925 cm-1三、炔烃类CH伸C≡C 伸CH弯(面内)CH弯(面外)~33002270~21001260~1245645~615~3.034.41~4.767.94~8.0315.50~16.25中中强四、取代苯类CH伸泛频峰骨架振动(CC=ν)CH弯(面内)CH弯(面外)3100~30002000~16671600±201500±251580±101450±201250~1000910~6653.23~3.335.00~6.006.25±0.086.67±0.106.33±0.046.90±0.108.00~10.0010.99~15.03变弱强三、四个峰,特征确定取代位置单取代邻双取代间双取代对双取代1,2,3,三取代1,3,5,三取代1,2,4,三取代﹡1,2,3,4四取代﹡1,2,4,5四取代﹡1,2,3,5四取代﹡五取代CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)770~730770~730810~750900~860860~800810~750874~835885~860860~800860~800860~800865~810~86012.99~13.7012.99~13.7012.35~13.3311.12~11.6311.63~12.5012.35~13.3311.44~11.9811.30~11.6311.63~12.5011.63~12.5011.63~12.5011.56~12.35~11.63极强极强极强中极强强强中强强强强强五个相邻氢四个相邻氢三个相邻氢一个氢(次要)二个相邻氢三个相邻氢与间双易混一个氢一个氢二个相邻氢二个相邻氢一个氢一个氢一个氢波波配体和16M n:通过比较配体与配合物的红外光谱图(附录图1.3)发现配体在1626.5 cm−1、1624.1 cm−1处有吸收峰,可归属为羰基C=O的伸缩振动,而图中3260.8 cm−1、3042 cm−1处的吸收峰可归属为酰胺基的ν(N-H)伸缩振动,2709 cm−1处的吸收峰归属为酚羟基ν(O–H)伸缩振动,1580.1 cm−1处的吸收峰归属为丁烯中C=C的特征吸收峰,1487.3 cm−1处的吸收峰为–C=N–N=C–基团产生[16-17]。

红外光谱特征峰解析常识红外光谱是一种非常常用的分析技术,它可以用于确定化合物的结构和功能团,检测物质的组分和纯度,因此在化学、药学、生物学、环境科学等领域中得到了广泛的应用。
在红外光谱中,各个峰的位置和强度可以提供有关样品中化学键的信息,因此对红外光谱中常见的峰有一些基本的了解是很重要的。
1. 对称振动(伸缩)峰:对称振动是指分子中的原子以相对同样的方式沿着键轴向两个方向振动。
这种振动形成了红外光谱中的峰。
一般来说,对称伸缩振动的峰位于4000-2500 cm-1的高频区域。
它们的强度通常比较强,因为对称振动会导致比较大的偶极矩的变化。
2. 非对称振动(伸缩)峰:非对称振动是指分子中的原子以不同的方式沿着键轴向两个方向振动。
非对称振动一般出现在4000-1500 cm-1的区域。
它们的强度通常比较弱,因为非对称振动会导致较小的偶极矩的变化。
3. 弯曲振动峰:分子中的原子围绕键的轴线进行弯曲振动,形成了红外光谱中的弯曲振动峰。
这些峰通常位于1500-400 cm-1的区域。
弯曲振动的强度通常非常弱,并且其强度与非对称伸缩振动的强度相比要弱得多。
4. 指纹区域峰:指纹区域是位于1500-400 cm-1的区域,其中包含了分子结构中独特的振动模式。
这些峰的位置和形状具有高度的特异性和指示性,可以用于确定物质的结构和识别化合物。
5.进一步解析峰的位置:了解常见的波数峰值范围和化学键的振动模式是很重要的,但要对红外光谱中的峰进行更准确的解析,通常需要参考红外光谱数据库或文献中的标准光谱。
这些数据库和文献中提供了大量的已知化合物的红外光谱数据,可以用来对未知样品进行鉴定。
总之,红外光谱分析是一种非常有用的技术,可以提供关于化合物结构和功能团的重要信息。
掌握常见的红外光谱特征峰的解析常识可以帮助科学家们更好地理解和利用红外光谱技术。

红外测试峰值对照图 WEIHUA system office room 【WEIHUA 16H-WEIHUA WEIHUA8Q8-主要基团的红外特征吸收峰基团振动类型波数(cm-1)波长(μm)强度备注一、烷烃类CH伸CH伸(反称)CH伸(对称)CH弯(面内)C-C伸3000~28432972~28802882~28431490~13501250~1140~~~~~中、强中、强中、强分为反称与对称二、烯烃类CH伸C=C伸CH弯(面内)CH弯(面外)单取代双取代顺式反式3100~30001695~16301430~12901010~650995~985910~905730~650980~965~~~~~~~~中、弱中强强强强强C=C=C为2000~1925 cm-1三、炔烃类CH伸C≡C 伸CH弯(面内)CH弯(面外)~33002270~21001260~1245645~615~~~~中中强四、取代苯类CH伸泛频峰骨架振动(CC=ν)CH弯(面内)CH弯(面外)3100~30002000~16671600±201500±251580±101450±201250~1000910~665~~±±±±~~变弱强三、四个峰,特征确定取代位置单取代邻双取代间双取代对双取代1,2,3,三取代1,3,5,三取代1,2,4,三取代﹡1,2,3,4四取代﹡1,2,4,5四取代﹡1,2,3,5四取代﹡五取代CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)770~730770~730810~750900~860860~800810~750874~835885~860860~800860~800860~800865~810~860~~~~~~~~~~~~~极强极强极强中极强强强中强强强强强五个相邻氢四个相邻氢三个相邻氢一个氢(次要)二个相邻氢三个相邻氢与间双易混一个氢一个氢二个相邻氢二个相邻氢一个氢一个氢一个氢五、醇类、酚类OH伸OH弯(面内)C—O伸O—H弯(面外)3700~32001410~12601260~1000750~650~~~~变弱强强液态有此峰波波配体和16M n:通过比较配体与配合物的红外光谱图(附录图发现配体在cm1、cm?1处有吸收峰,可归属为羰基C=O的伸缩振动,而图中cm?1、3042 cm1处的吸收峰可归属为酰胺基的ν(N-H)伸缩振动,2709 cm?1处的吸收峰归属为酚羟基ν(O–H)伸缩振动,cm1处的吸收峰归属为丁烯中C=C的特征吸收峰, cm1处的吸收峰为–C=N–N=C–基团产生[16-17]。

如何进行红外光谱解析红外光谱解析是一种广泛应用于化学、生物、材料科学等领域的测试技术,通过分析物质在红外光波段的吸收和散射特性,可以获得物质的结构信息、成分组成以及其他相关性质。
本文将介绍红外光谱解析的基本原理、实验操作步骤以及数据分析方法,帮助读者了解如何进行红外光谱解析。
一、基本原理红外光谱解析的基本原理是物质分子在吸收红外光时,会发生振动和转动,并发生状态之间的转变。
这些振动和转动产生的谐振频率,与分子内部的键长、键角等结构参数有关,因此可以通过测量红外光谱图谱来了解物质的结构特征。
二、实验操作步骤1. 仪器准备:将红外光谱仪连接电源并打开。
根据待测物的性质,选择适当的样品盒(液态或固态)和检测模式(透射或反射)。
2. 样品处理:对于液态样品,取少量样品加入透射池中,移除气泡并将其密封;对于固态样品,将样品压制成片或粉碎并放置在反射盒中。
3. 启动仪器:根据仪器操作手册,进行光谱仪的启动和样品检测参数的设置。
4. 开始检测:点击仪器软件上的“开始”按钮,红外光谱仪开始发送红外光,并通过探测器接收返回的信号。
5. 数据采集:红外光谱仪会将接收到的信号转化为电信号,并通过数据采集软件记录下来。
采集过程通常需要数秒至数分钟。
6. 数据处理:获取红外光谱图谱后,使用特定的数据处理软件进行谱图展示和数据分析。
三、数据分析方法1. 谱图展示:使用数据处理软件将红外光谱图谱进行展示,在横轴上表示波数,纵轴表示吸收强度。
确保谱图的分辨率和信噪比足够高,以保证后续的数据分析准确性。
2. 峰值鉴定:根据谱图上的吸收峰,确定物质的各种官能团或键的存在。
通过比对已知物质的红外光谱数据库,寻找吸收峰的对应官能团或键。
3. 定量分析:利用谱图上的吸收峰的强度,可以进行物质的定量分析。
通过校正曲线或比色法等方法,计算物质的浓度或含量。
4. 结构确定:根据红外吸收峰的波数和强度,可以获得物质的结构信息。
通过对比不同官能团或键的红外吸收谱图,推测和确认物质的结构特征。

主要基团的红外特征吸收峰反式985 910~905 730~650 980~965三、炔烃类CH伸C≡C 伸CH弯(面内)CH弯(面外)~33002270~21001260~1245645~615~~~~中中强四、取代苯类CH伸泛频峰骨架振动(CC=ν)CH弯(面内)CH弯(面外)3100~30002000~16671600±201500±251580±101450±201250~1000~~±±±±~~变弱强三、四个峰,特征确定取代位置910~665单取代邻双取代间双取代对双取代1,2,3,三取代1,3,5,三取代1,2,4,三取代﹡1,2,3,4四取代﹡1,2,4,5四取代﹡1,2,3,5四取代﹡五取代CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)CH弯(面外)770~730770~730810~750900~860860~800810~750874~835885~860860~800860~800860~800865~810~860~~~~~~~~~~~~~极强极强极强中极强强强中强强强强强五个相邻氢四个相邻氢三个相邻氢一个氢(次要)二个相邻氢三个相邻氢与间双易混一个氢一个氢二个相邻氢二个相邻氢一个氢一个氢一个氢五、醇类、酚类OH伸OH弯(面内)3700~3200~~变弱液态有此峰波波配体和16Mn:通过比较配体与配合物的红外光谱图(附录图发现配体在 cm1、 cm?1处有吸收峰,可归属为羰基C=O的伸缩振动,而图中 cm1、3042 cm?1处的吸收峰可归属为酰胺基的ν(N-H)伸缩振动,2709 cm1处的吸收峰归属为酚羟基ν(O–H)伸缩振动, cm?1处的吸收峰归属为丁烯中C=C的特征吸收峰, cm1处的吸收峰为–C=N–N=C–基团产生[16-17]。
红外光谱分峰拟合
红外光谱分峰拟合是一种分析红外光谱图像的方法,用于确定样品中存在的不同分子或官能团的类型和数量。
下面是红外光谱分峰拟合的基本步骤:
1.数据预处理:将原始红外光谱数据进行预处理,例如进行峰值对齐、基线校正和去噪等处理。
2.初步拟合:使用高斯或洛仑兹函数对谱图进行初步拟合,以确定潜在的谱峰位置。
3.定义拟合函数:根据样品的化学特性和红外光谱数据,选择合适的拟合函数,例如高斯或洛仑兹函数等。
4.拟合参数设置:设置拟合函数的参数,包括峰形、峰位、峰高等参数。
5.拟合优化:通过最小二乘法等方法对拟合函数进行优化,以获得最佳的拟合结果。
6.分析谱峰:对拟合结果进行分析,确定样品中存在的分子或官能团的类型和数量。
7.结果评估:根据分析结果进行结果评估,检查拟合效果和确定的峰位是否准确。
8.红外光谱分峰拟合是一种常用的谱图分析方法,可以帮助科学家们深入研究不同化合物或材料的化学结构和性质,为材料研究、化学分析等领域的应用提供重要的支持。
傅里叶红外光谱分析第一节一般原理电子能级跃迁所产生的吸收光谱,主要在近紫外区和可见区,称为可见-紫外光谱;键振动能级跃迁所产生的吸收光谱,主要在中红外区,称为红外光谱;自旋的原子核在外加磁场中可吸收无线电波而引起能级的跃迁,所产生的吸收光谱称为核磁共振谱。
第二节紫外光谱一、紫外光谱的基本原理用波长范围200 nm~800 nm的光照射含有共轭体系的的不饱和化合物的稀溶液时,部分波长的光被吸收,被吸收光的波长和强度取决于不饱和化合物的结构。
以波长l为横座标,吸收度A为纵座标作图,得紫外光谱,或称电子光谱。
是化合物紫外光谱的特征常数。
紫外光谱中化合物的最大吸收波长λmax可见-紫外光谱适用于分析分子中具有π键不饱和结构的化合物。
二、紫外光谱在有机结构分析中的应用随着共轭体系的延长,紫外吸收向长波方向移动,且强度增大(π→π*),因此可判断分子中共轭的程度。
利用紫外光谱可以测定化合物的纯度或含量。
第三节红外光谱一、红外光谱的基本原理用不断改变波长的红外光照射样品,当某一波长的频率刚好与分子中某一化学键的振动频率相同时,分子就会吸收红外光,产生吸收峰。
用波长(λ)或波长的倒数—波数(cm-1)为横坐标,百分透光率(T%)或吸收度(A)为纵坐标做图,得到红外吸收光谱图(IR)。
分子振动所需能量对应波数范围在400 cm-1~4000 cm-1。
二、红外吸收峰的位置和强度分子中的一个化学键可有几种不同的振动形式,而产生不同的红外吸收峰,键的振动分为两大类。
伸缩振动,用n表示,原子间沿键轴方向伸长或缩短。
弯曲振动用δ表示,形成化学键的两个原子之一与键轴垂直方向作上下或左右弯曲。
组成化学键的原子的质量越小,键能越高,键长越短,振动所需能量越大,吸收峰所在的波数就越高。
红外光谱的吸收峰分为两大区域:4000 cm-1~1330 cm-1区域:特征谱带区,是红外光谱分析的主要依据。
1330 cm-1~650 cm-1区域:指纹区。
红外光谱分析(FT-IR)傅立叶变换红外光谱(FT-IR)是一种强大的技术,可用于获取吸收/排放固体、液体或气体的红外光谱。
当红外辐射穿过被测样品时,一部分红外辐射会被官能团的特定共价键吸收,另一部分红外辐射则直接穿透收集到的光谱代表了分子的吸收和传输,形成了用于化学鉴定的分子指纹。
这也使得红外光谱可用于多种类型的分析。
傅立叶变换红外光谱仪同时收集宽波长范围内的高分辨率光谱,这与色散光谱仪相比具有显著的优势,色散光谱仪一次只能测量相当窄波长范围内的峰值强度。
傅立叶变换红外光谱(FT-IR)分析。
傅立叶变换红外光谱仪可用于所有使用色散仪来提高灵敏度和速度的应用,能够优于红外光谱分析的色散法或滤光片法取决于其:1,非破坏性;2,无需外部校准;3,速度更快;4,灵敏度更高;5,光通量更高;6,操作更简单。
傅立叶变换红外光谱仪分析应用。
1.基于同质异性、同系物、几何和光学异构体的光谱差异进行化学鉴定;2.根据吸收的波长鉴定被测化学品中的官能团;3.通过研究潜在污染物的峰值进行纯度估算;4.通过比较特定官能团的峰跟踪化学反应过程;5.通过监测特定峰对化学物质进行定量分析。
百泰派克生物科技BTP基于CNAS/ISO9001双重质量认证体系建立七大检测平台,采用Thermo公司Nicolet系列仪器建立FT-IR分析平台,测定样品中蛋白和多肽的红外光谱,并进行后续的基线校正、Gaussian去卷积、二阶导数拟合,最终根据峰面积确定样品中蛋白和多肽的二级结构信息。
联系我们,免费项目咨询。
百泰派克生物科技生物制品表征服务内容。
FT-IR分析一站式服务。
您只需下单-寄送样品。
百泰派克生物科技一站式服务完成:样品处理-上机分析-数据分析-项目报告。
origin读红外光谱的峰值
根据定义,红外光谱是通过检测被样品吸收或发射的红外辐射来进行分析的技术。
对于红外光谱的峰值,其实是指样品在红外波长范围内的吸收峰(或发射峰)。
在红外光谱中,不同化学物质具有不同的分子结构和键,导致吸收红外辐射的方式也不同。
这些吸收或发射谱线(峰值)的位置和强度与样品的化学成分和相对浓度有关。
为了获得红外光谱的峰值,通常需要进行以下步骤:
1. 准备样品:将待测样品制备成适合红外光谱分析的形式,例如液体样品可以通过将其放置在透明的红外吸收容器中适当稀释,固体样品可以采用如固样法、压片法等方法。
2. 选择适当的仪器:常用的红外光谱仪包括傅里叶变换红外光谱仪(FTIR)和红外分光光度计(IR Spectrometer),根据需要选择合适的仪器。
3. 进行测量:将样品放置在红外光谱仪中,设置适当的参数(如扫描范围、积分时间等),开始测量。
在红外光谱仪中,红外辐射通过样品并根据样品的吸收或发射特性发生变化,变化后的辐射通过仪器进行检测和记录。
4. 解析数据:通过对测定得到的数据进行处理和解析,可以得到样品的红外光谱图。
在红外光谱图中,可以识别出吸收或发射谱线,并确定其位置和强度,即峰值。
需要注意的是,红外光谱技术是一种无损分析方法,适用于各种物质的分析,尤其在有机化学、无机化学、生物化学等领域
具有广泛的应用。
不同的峰值可以提供有关样品化学元素、官能团等信息,从而用于分析和鉴定物质的结构和性质。
红外光谱分析
第一节吸收光谱的一般原理
电子能级跃迁所产生的吸收光谱,主要在近紫外区和可见区,称为可见-紫外光谱;键振动能级跃迁所产生的吸收光谱,主要在中红外区,称为红外光谱;自旋的原子核在外加磁场中可吸收无线电波而引起能级的跃迁,所产生的吸收光谱称为核磁共振谱。
第二节紫外光谱
一、紫外光谱的基本原理
用波长范围200 nm~800 nm的光照射含有共轭体系的的不饱和化合物的稀溶液时,部分波长的光被吸收,被吸收光的波长和强度取决于不饱和化合物的结构。
以波长l为横座标,吸收度A为纵座标作图,得紫外光谱,或称电子光谱。
是化合物紫外光谱的特征常数。
紫外光谱中化合物的最大吸收波长λ
max
可见-紫外光谱适用于分析分子中具有π键不饱和结构的化合物。
二、紫外光谱在有机结构分析中的应用
随着共轭体系的延长,紫外吸收向长波方向移动,且强度增大(π→π*),因此可判断分子中共轭的程度。
利用紫外光谱可以测定化合物的纯度或含量。
第三节红外光谱
一、红外光谱的基本原理
用不断改变波长的红外光照射样品,当某一波长的频率刚好与分子中某一化学键的振动频率相同时,分子就会吸收红外光,产生吸收峰。
用波长(λ)或波长的倒数—波数(cm-1)为横坐标,百分透光率(T%)或吸收度(A)为纵坐标做图,得到红外吸收光谱图(IR)。
分子振动所需能量对应波数范围在400 cm-1~4000 cm-1。
二、红外吸收峰的位置和强度
分子中的一个化学键可有几种不同的振动形式,而产生不同的红外吸收峰,键的振动分为两大类。
伸缩振动,用n表示,原子间沿键轴方向伸长或缩短。
弯曲振动用δ表示,形成化学键的两个原子之一与键轴垂直方向作上下或左右弯曲。
组成化学键的原子的质量越小,键能越高,键长越短,振动所需能量越大,吸收峰所在的波数就越高。
红外光谱的吸收峰分为两大区域:
4000 cm-1~1330 cm-1区域:特征谱带区,是红外光谱分析的主要依据。
1330 cm-1~650 cm-1区域:指纹区。
每一化合物在指纹区都有它自己的特征光谱,对分子结构的鉴定能提供重要信息。
(很强);s(强);m(中强);w(弱);红外吸收峰的强弱用下列符号表示:v
s
v
(很弱);b(宽峰)。
w
凡能使键增强的因素,引起峰位向高波数方向移动,反之,则向低波数方向移动。
三、各类化合物的红外光谱举例
(一)烃类化合物
注:烷烃,即饱和烃,是只有碳碳单键和碳氢键的链烃。
烷烃的通式为CnH2n+2。
烯烃是指含有C=C键(碳-碳双键)(烯键)的碳氢化合物,单链烯烃分子通式为CnH2n 炔烃,为分子中含有碳碳三键的碳氢化合物的总称,其官能团为碳-碳三键(C≡C),分子通式为CnH2n-2
烯烃的C=C伸缩振动n(C=C)吸收峰在1640 cm-1~1680 cm-1处;
炔烃的C≡C伸缩振动n(C≡C)吸收峰在2100 cm-1~2200 cm-1处。
但当取代烯烃或炔烃相当对称时,n(C=C)或n(C≡C)不会引起偶极矩的变化而不出现吸收峰。
C-H键伸缩振动所产生的吸收峰在光谱的高频区。
烷烃中n(C
sp3
-H)在2800 cm-1~
3000 cm-1,烯烃的n(C
sp2-H)在3000 cm-1~3100 cm-1,而炔烃的n(C
sp
-H)在
3300 cm-1,据此可区别饱和和不饱和烃。
各种C-H键的弯曲振动所产生的吸收峰
在光谱的低频区,烷烃中-CH
3的δC-H约1450cm-1(m)和1380cm-1(w),-CH
2
的δC-H
约为1450cm-1(m),如果约在1380cm-1出现等强的双峰,表明有异丙基存在。
烯烃的δC-H有面内和面外两种,烯烃的结构不同,吸收峰的位置存在差别,这对烯烃异构体的鉴别,可提供有价值的信息。
芳环的C-H键伸缩振动n(C-H)在3000 cm-1~3100 cm-1(m);C-C键的伸缩振动n(C=C)在1500 cm-1~1600 cm-1,芳烃的骨架振动一般在1500 cm-1和1600 cm-1处,最多可能出现强度不等的4个峰,这是区别于烯烃n(C=C)的重要特征;芳烃C-H键的面外弯曲在680 cm-1~880 cm-1。
这些吸收峰根据苯环上取代基的个数和位置不同而发生变化。
因此,该区域可作为判别取代苯异构体的依据。
取代苯的弯曲振动特征吸收频率
(二)含氧化合物
醇类或酚类化合物的红外特征吸收峰是在3300 cm-1左右有一个宽而强的n
O-H
吸收带,可作为区别醇、酚与醚的重要依据。
醛和酮的C=O特征吸收峰在1725 cm-1左右。
醛C=O上的C-H的伸缩振动在2810 cm-1—2715 cm-1。
羧酸通常以二聚体形式存在,C=O伸缩振动在1720 cm-1左右,而O-H的吸收在3300 cm-1~2500 cm-1间有一个相当宽的峰,中心位于3000 cm-1,因此C-H 的伸缩振动吸收峰常被覆盖。
(三)胺类化合物
胺类化合物的特征吸收峰是在3200 cm-1~3500cm-1范围内有N-H的伸缩振动,伯胺有两个吸收峰,仲胺有一个吸收峰,而叔胺在此范围内无吸收峰。
四、红外光谱的解析
利用红外图谱解析有机化合物的结构,首先根据官能团区域中的特征吸收峰的位置,判别可能存在什么官能团;然后找出该官能团的相关峰,以确证该官能团的存在;最后,将所测图谱与标准图谱对照。
第四节核磁共振谱
一、核磁共振基本原理
当外界提供的能量等于两种自旋取向的能级差(△E)时,核从低能级自旋状态向高能级自旋状态跃迁,即发生核磁共振(NMR)。
广泛研究的是1H和13C的核磁共振谱即1HNMR和13CNMR。
二、化学位移
(一)屏蔽作用
核外的电子云密度越高,则受到的屏蔽作用就越强,共振时所需的外加磁场强度也越高,其信号必然在较高磁场出现。
反之,核外的电子云密度越低,信号必然在较低磁场出现。
(二)化学位移
因质子所处的化学环境不同,即核外电子云密度不同,所受屏蔽作用的强弱不同而引起质子在核磁共振谱中吸收信号位置的移动,称为化学位移。
一般采用四甲基硅烷(TMS)为参考物。
用δ表示化学位移值时,规定TMS的δ值为0.0,TMS信号左侧的δ值为正,右侧的δ值为负。
一般化学位移值在0.0~14.0范围内。
在核磁共振谱中,信号的数目表示一个分子中有几种类型的H原子。
1、化学位移d值随着其邻近原子(或基团)中电负性增强而增大。
2、烃基氢的d值为芳环上氢>烯基氢>炔基氢>饱和碳原子上的氢。
3、饱和碳原子上的氢的d值为3°>2°>1°。
、-SH等活泼氢,由于氢键的形成使d值向低场位移,并随4、-OH、-COOH、-NH
2
测定时的温度、浓度而变化。
这些质子由于溶剂效应,其d值可能随测定时的溶剂不同而不同。
三、吸收峰的面积——质子的数目
各类质子的数目与其产生的信号强度有关。
四、自旋偶合-裂分
相邻的不等性质子由于自旋而产生的磁性间相互作用,称为自旋-自旋偶合。
自旋偶合所引起的信号吸收峰裂分而峰增多的现象,称为自旋-自旋裂分。
质子吸收信号中各裂分峰间的距离称为偶合常数,以J表示,单位为Hz。
J值越大,核间自旋-自旋偶合的作用越强。
相互偶合的两组信号具有相同的偶合常数。
偶合常数只依赖于邻近质子的自旋偶合作用,而与外加磁场强度无关。
(1)自旋偶合主要发生在同一碳或相邻碳上的不等性质子之间。
:
(2)活泼质子一般图谱中观察不到相互自旋偶合作用。
(3)当一组质子相邻的同类质子数为n时,该组质子的信号裂分为(n+1)重峰,称之为(n+1)规律。
一般用s表示单峰,d表示二重峰,t表示三重峰,q表示四重峰,m表示四重峰以上的多重峰。
(4) 各裂分峰的强度比等于二项式(a+b)n展开式的各项系数,n为邻接氢质子的数目。
五、1HNMR谱的解析
解析核磁共振谱,主要是从其中寻找信号的位置、数目、强度及裂分情况的信息。
从信号的数目可知分子中有多少种不同类型的质子;从信号的位置(即化学位移δ值)可知每类质子的电子环境;各吸收峰占有的相对面积则表示各类质子的相对数目;从信号的裂分情况可提供邻近基团结构的信息。
如果再结合红外光谱和其他光谱,就可推测化合物的结构。
对于某些已知分子式的简单化合物,只需它的1HNMR谱就可推测其结构。
将IR 谱与1HNMR谱结合应用,从前者可知所测化合物的官能团(烯基、羟基、羰基等),从后者则能得知化合物的结构。
对于结构复杂的化合物,特别是确定天然有机化合物的结构,往往需要将
1HNMR谱与 IR、MS、UV、13CNMR谱等结合起来分析。