液相法制备纳米颗粒的机制.
- 格式:doc
- 大小:584.01 KB
- 文档页数:19

液相制备纳米材料的原理、方法、形成机理和结构液相法实在液体状态下通过化学反应制取纳米材料方法的总称,又称为湿化学法或溶液法。
现在,有各种各样的制备方法,文献中无公认一致的分类方法,相反还有些凌乱。
为清晰醒目,特点明显,便于理解。
这里将液相材料的纳米制备方法分为:沉淀法、溶胶-凝胶(sol-gel)法、水热法、化学还原法、化学热分解法、微乳胶法、声化学法、电化学法和水中放电法等中。
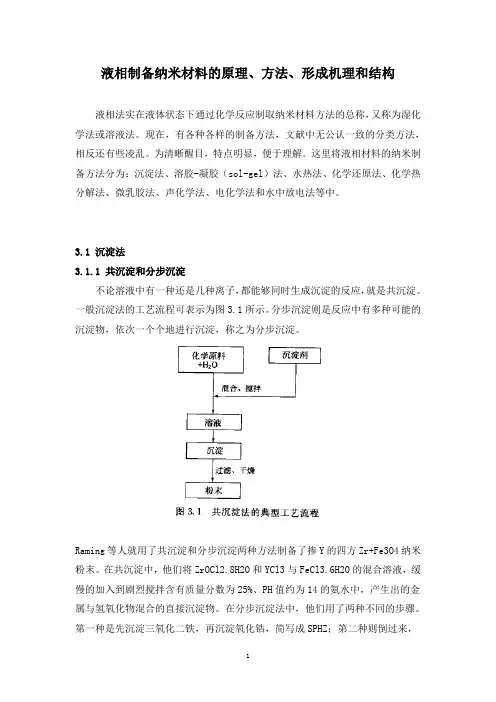
3.1 沉淀法3.1.1 共沉淀和分步沉淀不论溶液中有一种还是几种离子,都能够同时生成沉淀的反应,就是共沉淀。
一般沉淀法的工艺流程可表示为图3.1所示。
分步沉淀则是反应中有多种可能的沉淀物,依次一个个地进行沉淀,称之为分步沉淀。
Raming等人就用了共沉淀和分步沉淀两种方法制备了掺Y的四方Zr+Fe3O4纳米粉末。
在共沉淀中,他们将ZrOCl2.8H2O和YCl3与FeCl3.6H2O的混合溶液,缓慢的加入到剧烈搅拌含有质量分数为25%、PH值约为14的氨水中,产生出的金属与氢氧化物混合的直接沉淀物。
在分步沉淀法中,他们用了两种不同的步骤。
第一种是先沉淀三氧化二铁,再沉淀氧化锆,简写成SPHZ;第二种则倒过来,先沉淀氧化锆,再沉淀氧化铁,简写成SPZH。
第一种具体操作是,将八水氯酸锆和YCl3的水溶液加到碱性的悬浮着氧化铁粒子的溶液中,因此是先沉淀氧化铁,再沉淀氧化锆。
第二种分步沉淀则是将六水氯化铁水溶液加到悬浮有氧化锆粉末粒子的碱性溶液中,因此是先沉淀氧化锆,再沉淀氧化铁。
这两种分步沉淀中,都是在剧烈搅拌中,将酸性的金属离子加入到碱性的氨水中,在悬浮液中导致金属氢氧化物的爆炸式成核。
经水洗后,在100℃下干燥成胶状,再在500-700℃温度内煅烧2h,以得到完全的晶体物质。
3.1.2 均匀沉淀无论是在CP还是在SP中,由于沉淀剂在金属溶液中的加入,哪怕是沉淀剂加入量很少,并不断的搅拌,在局部溶液中的沉淀剂浓度都可以变得很高,于是这些地方就首先沉淀,使沉淀变得不均匀,必须在溶液中消除不均匀的沉淀,而使整个溶液中均匀的生成沉淀。


纳米功能材料的制备与表征近年来,纳米技术发展日新月异,纳米材料的制备与应用也得到了广泛的关注。
纳米功能材料的制备与表征是纳米科技中不可缺少的环节,在纳米科技的各个领域中都有着重要的应用。
今天,我们就一起来了解一下纳米功能材料的制备与表征的相关知识。
一、纳米功能材料的制备方法在制备纳米功能材料时,通常需要通过一些特殊的方法来实现纳米级精度。
其中,主要有以下几种方法:1. 物理制备方法物理制备方法是指通过物理手段来制造纳米材料,主要包括机械法、热处理法、蒸发法、溅射法等。
机械法是指通过机械力将材料切割成纳米级别的微粒。
常用的机械制备方法有球磨法、流化床法等。
热处理法是指将材料在高温下进行一系列的热处理,使其形成纳米级别的颗粒。
常用的热处理方法有高温还原法、热分解法等。
蒸发法是指将材料在真空条件下蒸发成薄膜,然后使用一些特殊的手段将其压缩成纳米级别的颗粒。
常用的蒸发法有电子束蒸发法、磁控溅射法等。
溅射法是指将材料放置在真空室中,在电子束或离子束的轰击下,使其形成纳米级别的颗粒。
常用的溅射法有磁控溅射法、光致发光溅射法等。
2. 化学制备方法化学制备方法是指通过化学反应来制备纳米材料,主要包括沉淀法、胶体溶胶法、微乳液法等。
沉淀法是指通过化学反应将材料溶液中的金属离子还原成金属颗粒,形成纳米级别的粒子。
常用的沉淀法有化学沉淀法、共沉淀法等。
胶体溶胶法是指在液相中制备纳米颗粒,主要通过控制反应条件来控制颗粒的大小和形态。
常用的胶体溶胶法有溶胶凝胶法、微乳液法等。
微乳液法是指在反应体系中加入表面活性剂,形成微胶团来控制粒子的大小和形态。
常用的微乳液法有水合胶体微乳液法、反应交替微乳液法等。
二、纳米功能材料的表征方法在研究纳米材料的表征时,常采用一些特殊的方法来观察其物理化学性质和结构特征。
其中,主要采用以下几种方法:1. 电子显微镜电子显微镜是一种用来观察纳米材料的表面形貌和结构的仪器。
主要包括扫描电子显微镜(SEM)、透射电子显微镜(TEM)等。







CHEMICAL INDUSTRY AND ENGINEERING PROGRESS 2012年第31卷第7期・1542・化工进展化学液相还原法制备零价铁纳米颗粒研究进展及展望樊明德1,2,袁鹏3,何宏平3,陈天虎4,朱建喜3,刘冬3,郝娇1(1内蒙古大学环境与资源学院,内蒙古呼和浩特 010021;2中国科学院广州地球化学研究所,矿物学与成矿学重点实验室,广东广州 510640;3中国科学院广州地球化学研究所,广东广州 510640;4合肥工业大学资源与环境工程学院,安徽合肥 230009)摘要:零价铁纳米颗粒磁性能卓越应用潜力巨大,已受到广泛关注。
本文综述了采用化学液相还原法制备纳米铁的研究进展。
总结了纳米铁制备过程中容易团聚和氧化两个关键问题:使用稳定剂可降低纳米铁团聚程度,表面包覆外壳可抑制纳米铁深度氧化。
并详细介绍了水合肼、多元醇、碱金属硼氢化物3种常用还原剂的还原性能及其在制备过程中表现出的优缺点。
提出化学液相还原制备纳米铁技术的发展依赖于对稳定剂与包覆剂的深入研究,对于还原反应工艺流程的工业化放大以及如何降低成本。
关键词:化学液相还原;零价铁纳米颗粒;水合肼;多元醇;碱金属硼氢化物中图分类号:O 614.81+1 文献标志码:A 文章编号:1000–6613(2012)07–1542–08 Review and prospect of zerovalent iron nanoparticles synthesized bychemical solution reduction processF AN Mingde1,2,YUAN Peng3,HE Hongping3,CHEN Tianhu4,ZHU Jianxi3,LIU Dong3,HAO Jiao1(1 School of Environment and Resources,Inner Mongolia University,Hohhot 010021,Inner Mongolia,China;2CAS Key Laboratory of Mineralogy and Metallogeny,Guangzhou Institute of Geochemistry,Chinese Academy of Sciences,Guangzhou 510460,Guangdong,China;3Guangzhou Institute of Geochemistry,Chinese Academy of Sciences,Guangzhou 510460,Guangdong,China;4School of Resources and Environmental Engineering,Hefei University of Technology,Hefei 230009,Anhui,China)Abstract:Zerovalent iron nanoparticles (ZVINs) have attracted much attention for their excellent magnetic properties and great potential in many practical applications. This review summarizes the details of synthesizing ZVINs by chemical reduction of iron salts in aqueous solution. ZVINs are easy to agglomerate and oxidize,which makes them difficult to prepare,study,and utilize. Agglomeration of ZVINs can be largely inhibited by stabilizing them with various dispersing agents and oxidation of ZVINs can be minimized by coating them with different shells. In the chemical solution reduction process,three kinds of reducing agents of hydrazine hydrate and polyols and alkali metal borohydrides with different reduction performance are often used to synthesize ZVINs. The advantages and disadvantages of these reducing agents for synthesizing ZVINs are discussed. Further developments of the chemical solution reduction process,to a great extent,depend on the insight into the behavior of dispersing agents and coated shells,on the industrial scale-up of the chemical reduction process,and on the low-cost preparation of ZVINs.收稿日期:2011-12-21;修改稿日期:2012-03-23。
纳米颗粒的化学制备方法纳米颗粒的各种化学制备方法及例举本文通过查阅图书馆中文数据库(CNKI)和外文数据库(Elsevier)相关资料,对纳米粒子的化学制备方法,如:沉淀法、溶胶-凝胶法、溶液蒸发法、化学气相沉积法和模板合成法等分别进行了举例说明,并对其各种化学制备方法的基本原理、化学反应及制备过程进行了简要的描述。
一.沉淀法1、共沉淀法Fe3O4磁性纳米粒子的共沉淀法制备研究陈亭汝青岛大学化学化工与环境学院孙瑾烟台南山学院以液相共沉淀法制备纳米磁性Fe3O4粒子的工艺,研究了反应搅拌速度、n(Fe3+ ) /n(Fe2+)的比例、pH值和熟化温度对制备纳米Fe3O4粒子的影响,并利用透射电镜表征观察Fe3O4纳米粒子的形貌。
研究结果表明,在搅拌速度较快的情况下制备纳米级Fe3O4颗粒的最佳合成工艺条件为:n(Fe3+)/n(Fe2+)为1﹒8:1(摩尔比),熟化温度70 ℃,熟化时间30 m in以氨水作沉淀剂最佳pH值是9左右,可制得纯度较高,粒径小于10nmFe3O4磁性粒子。
(1)制备原理搅拌速度的影响纳米颗粒可以自动的进行团聚降低本身的能量,适当的搅拌速度可以破坏团聚体中小微粒之间的库仑力和范德华力,有利于纳米微粒在混合溶液中保持稳定和分散均匀。
由于搅拌速度的加快有利于反应物之间的充分接触,能避免搅拌不均而产生的局部浓度过高,使晶核生成和长大都均匀地进行,从而粒径小且分布均匀。
因此较高的搅拌速度有利于合成较小粒径的纳米粒子。
(2)试剂及反应方程式试剂:FeCl3*6H20, FeCl2*4H20, NH3*H20, NaOH,柠檬酸、尿素均为分析纯。
反应方程式采用液相共沉淀法制备纳米Fe3O4 的反应原理如下:Fe2+ + 2Fe3+ + 8OH-- =Fe3O4 +4H2O(3)制备工艺过程如下图2、均匀沉淀法均匀沉淀法合成纳米氧化铁欧延,邱晓滨,许宗祥,林敬东,廖代伟厦门大学物理化学研究所,化学系,固体表面物理化学国家重点实验室以尿素为均匀沉淀剂、氯化铁为原料,采用均匀沉淀法在不同的条件下合成具有实用价值的a型纳米氧化铁.用XRD和TEM测定产品的形貌并确定产品的纳米尺度.实验表明,所合成的Fe2O3为α型,粒径在20~40 nm范围,且分散性好.(1)制备原理采用均匀沉淀法,利用尿素高温发生水解反应(1)(如下),缓慢生成构晶离子,随着反应的缓慢进行,溶液的pH值逐渐上升.Fe3+和OH一反应,并在溶液的不同区域中均匀地形成铁黄粒子,尿素的分解速率直接影响了形成铁黄粒子的粒度,而尿素的分解速率又由反应温度所决定.温度很低时,离子具有的能量较低,晶粒生成速度很小,虽然有利于形成稳定的晶粒,但反应速度太慢,使得粒径大且分布不均匀.反应温度升高则反应速度加快,晶粒形成的速度也加快,但温度过高,一方面溶液的过饱和度下降,同时不利于形成稳定的晶粒,晶粒生成速度反而下降.(2)反应方程式(3)合成过程二.溶液蒸发法1.冷冻干燥法冷冻干燥法制备氧化铜纳米粉体的实验研究刘军东北大学机械工程与自动化学院徐成海沈阳大学师范学院利用冷冻干燥法,以无机化合物硫酸铜和氢氧化钠为原料,选取铜氨络合物为前驱体,制备出了粒径为20~50nm的氧化铜粉和带有均匀~10nm孔隙的多孔颗粒材料,并进行了TEM 和SEM检测。
钴基纳米合金颗粒的液相还原法制备钴铁合金具有较高的饱和磁感应强度、较高的居里温度以及较低的矫顽力,因而广泛应用于电子工业。
钴铁合金纳米颗粒的磁性与其颗粒的尺寸、形状有很强的相关性,所以通过一定的方法制备出不同尺寸,不同形貌的钴基纳米颗粒有着重要的科学和应用意义。
如今人们可以通过电弧熔炼,气相沉积,溅射等方法制备出钴基合金纳米颗粒,但这些方法存在产率低,装置复杂,生产成本高等缺点,这制约了钴基纳米颗粒的大规模生产应用。
液相还原法制备合金颗粒具有操作简单,条件温和,易于大规模生产的优势,因此在磁性金属纳米材料的制备中被广泛采用。
本论文通过改变反应温度、溶剂浓度、还原剂浓度、碱性条件、搅拌速度探讨形貌控制机制。
制备出了尺寸不同的Co2Fe纳米颗粒并对其形貌进行了表征。
另外,通过使用次亚磷酸钠作为还原剂,利用D-葡萄糖酸钠作为络合剂制备Co2FeSn纳米颗粒,并对产物的成分进行了表征。
关键词:钴铁合金,纳米颗粒,液相还原法第一章引言磁性材料具有能量转换、储存或改变能量状态的功能,是重要的功能材料。
目前,磁性材料广泛应用于信息记录存储装置如硬盘、磁带,电工产品如变压器,电动机,以及通信设备和各种电子产品中[1]。
钴铁合金及其颗粒具有较高的饱和磁感应强度以及较高的居里温度,探究不同变量对钴铁合金颗粒形貌的影响有着较高的科研和应用价值[2]。
同时Co2Fe是扩散法制备Co2FeSn前提,制备出具有一定形貌的Co2Fe是控制Co2FeSn形貌的关键。
然而由于以往的制备方法及工艺的要求较为严苛,限制了钴铁合金的大量应用,所以探索出一种成本低廉,操作简便的制备方法将极大地推动磁性材料的广泛应用。
液相还原法制备Co2Fe具有成本低廉、设备简单、条件温和的特点,因而本实验主要探索不同变量对Co2Fe 形貌的影响,以及利用液相还原法直接制备Co2FeSn。
1.1 纳米材料概述纳米科学技术被认为是21世纪非常重要的一门科学技术。
液相化学还原法制备纳米银颗粒化学还原法:运用化学试剂通过得失离子的方法进行化学反应的方法分散剂(Dispersant)是一种在分子内同时具有亲油性和亲水性两种相反性质的界面活性剂。
可均一分散那些难于溶解于液体的无机,有机颜料的固体颗粒,同时也能防止固体颗粒的沉降和凝聚,形成安定悬浮液所需的药剂纳米银作为一种贵金属纳米材料,具有比表面积大,表面活性高,导电性优异,催化性能良好等优点[1],在物理、化学、生物等方面具有显著的优势,包括表面增强拉曼散射[2]、导电[3]、催化[4]、传感[5]以及广谱抗菌活性[6]等。
近年来,纳米银的特殊性质被日益深入地了解,并在微电子材料[7]、催化材料、低温超导材料、电子浆料、电极材料[8]、光学材料、传感器等工业领域得到广泛应用,此外,其优良的抗菌性愈发受到人们的重视[9],成为新型功能材料的研究热点。
国内外关于纳米银的制备和可控性研究已经有了大量的报道[10],常用的制备方法包括水热法[11]、凝胶溶胶法、微乳液法[12]、模板法[13]、电还原法[14]、光还原法[15]、超声还原法[16]等化学还原法,以及球磨法、磁控溅射法等物理方法。
化学还原法由于其操作方便、设备简单、投入较少、可控性好,是实验室条件下主要的纳米银的制备方法。
然而,化学法制备纳米银也存在着一定的缺陷,热力学性质不稳定如比表面积大、表面能高等,从而影响纳米银的物理特性和功能。
本文拟利用聚乙烯吡咯烷酮(PVP)作为银的水相分散剂,在水热化学还原环境中,控制银晶体的生长,使之形成尺寸稳定的纳米微晶,通过改变PVP 的添加量与反应过程产物监控,研究PVP 对纳米银晶体成核生长的影响。
实验部分1.1.2 实验仪器T-1000 型电子天平,恒温磁力搅拌水浴,CQX25-06 型超声清洗仪,TGL-16 型高速离心机,DZF-6020 真空干燥箱,VIS-723 型分光光度计,产品形貌观察使用透射电子显微镜,D8 DISCOVER GADDS 型X 射线衍射仪。
纳米溶液的原理纳米溶液是一种将纳米颗粒分散在溶剂中形成的液体体系。
纳米颗粒的尺寸通常在1到100纳米之间,随着尺寸的减小,纳米颗粒的比表面积增大,因此在液相中具有较高的活性和特殊性质。
纳米溶液的原理涉及纳米颗粒分散和稳定的过程。
纳米颗粒的分散是指将纳米颗粒均匀地分散在溶剂中,避免其聚集和沉淀。
在溶液中,纳米颗粒可能会因为分子间相互吸引力而发生簇集。
为了使纳米颗粒分散均匀,需要采取一些分散剂或表面活性剂来阻止其簇集。
分散剂可以通过吸附在纳米颗粒的表面,形成电荷或静电斥力来防止颗粒的聚集。
此外,分散剂还可以改变溶剂的表面张力,使其与纳米颗粒更好地相容,从而增强颗粒的分散性。
纳米溶液的稳定性是指纳米颗粒在溶液中长时间保持分散状态的能力。
稳定性的实现需要考虑多种因素,如颗粒间的静电斥力、范德瓦尔斯力、电双层作用、溶剂介质的性质等。
其中,静电斥力是纳米颗粒稳定性的主要因素之一。
当纳米颗粒表面带有电荷时,它们会相互排斥,阻止颗粒的聚集。
电双层作用是指溶液中带电颗粒表面附近存在一个电荷层,通过该层可以实现颗粒间的斥力。
范德瓦尔斯力是吸引力,当纳米颗粒之间的距离足够近时,经典的范德瓦尔斯力会导致颗粒的聚集和沉淀。
为了增加纳米颗粒的稳定性,可以通过控制颗粒的表面电荷密度、调节溶液的pH值、添加分散剂和控制环境温度等方式来改变纳米颗粒之间相互作用的力。
纳米溶液的原理还涉及到纳米颗粒的制备方法。
常见的制备方法包括湿法合成和气相沉积。
湿法合成是通过在溶液中加入适当的前驱体,然后通过控制反应条件(如温度、pH值)来合成纳米颗粒。
气相沉积是将气体或气体的原料引入反应室中,通过化学反应生成纳米颗粒,然后通过控制反应条件和沉积基底表面的温度来控制纳米颗粒的生长。
纳米溶液在许多领域具有广泛的应用前景。
在生物医学领域,纳米溶液可以用于药物传递、诊断和治疗等方面。
由于纳米颗粒具有高比表面积和良好的生物相容性,可以将药物包裹在纳米颗粒表面或内部,实现药物的控释和靶向传递。
第43卷 第4期2007年7月 南京大学学报(自然科学)J OU RNAL OF NANJ IN G UN IV ERSIT Y(NA TU RAL SCIENCES)Vol.43,No.4J uly,2007环境科学专栏改进液相还原法制备纳米零价铁颗粒3高树梅1,王晓栋133,秦 良1,罗 斯1,赵 欣1,刘树深2,王连生1(1.污染控制与资源化研究国家重点实验室,南京大学环境学院,南京,210093;2.,同济大学环境科学与工程学院,上海,200092)摘 要: 纳米零价铁颗粒具有优越的吸附性能和很高的还原活性,因此在环境污染的处理和环境修复领域应用广泛.采用一种改进液相还原法制备纳米零价铁颗粒,通过添加高分子分散剂聚乙烯吡咯烷酮(PV P)和乙醇对纳米铁颗粒进行表面物理改性,从而达到改善其在水溶液中分散性的目的.实验过程中,机械搅拌条件下,将一定浓度的NaB H4水溶液(或乙醇2水混合溶液)迅速添加到一定浓度的FeSO4・7H2O水溶液(或乙醇2水混合溶液)中,短时间即可产生大量铁粉.过程无需氮气保护,反应迅速;采用透射电子显微镜(TEM),X射线衍射(XRD),比表面测定仪(B ET)三种方法对制得的纳米铁颗粒进行表征.TEM表征的结果表明:颗粒分散较均匀,粒径小,平均粒径为60nm(水溶液)和40nm (乙醇2水混合溶液),实验过程中添加的聚乙烯吡咯烷酮对颗粒的分散起到了很好的改善作用,其机理主要是通过分散剂吸附改变粒子的表面电荷分布,产生静电稳定效应,空间位阻作用和静电空间位阻稳定效应来达到分散效果;加入乙醇后,可能是由于乙醇中包含大量的自由的强极性羟基基团,在水溶液中这些基团与金属离子之间形成螯合键,紧密包覆在金属离子周围,形成一个有限制形状的有限结构,使合成的纳米粒子的大小被限制,从而达到改性的目的.XRD表征的结果表明:在扫描衍射角度(2θ)为30°~100°时,出现衍射峰时对应的2θ分别为45°、65°、82°左右,对照铁的标准PDF卡片发现,刚好对应相应的110晶面衍射(44.6732°),200晶面衍射(65.0211°),211晶面衍射(82.3326°),同时通过布拉格方程及电子衍射花样的分析,均表明颗粒为单质铁,没有出现氧化铁杂质、纯度高.采用B ET表征的结果表明:颗粒的比表面积为47.1m2/g(水溶液)和68.41m2/g(乙醇—水混合溶液),远远高于普通铁粉的比表面积.多次试验的结果表明:该方法工艺非常稳定.关键词: 纳 米,零价铁颗粒,制 备,液相还原法中图分类号: TM273 33基金项目:江苏省自然科学基金创新人才项目(BK200418),国家自然科学基金(20507008,20477018),国家973项目(2003CB415002)收稿日期:2007-02-12 通讯联系人,E2mail:wan gxd@Preparation of N ano Zero2valent Iron Particles by Modif iedLiquid Phase R eduction MethodGao S hu2Mei1,W ang X i ao2Dong1,Qi n L i ang1,L uo S i1,Zhao X i n1,L i u S hu2S hen2,W ang L i an2S heng1(1.State Key Laboratory of Pollution Control and Resource Reuse,School of Environment,Nanjing University,Nanjing,210093,China;2.Key Laboratory of Yangtze River Water Environment, Ministry of Education,College of Environmental Science and Engineering,Tongji University,Shanghai,200092,China)Abstract: Nano zero2valent iron particles are very absorptive and also highly reductive.So they are widely used in the environmental restoration and the treatment of the environmental contaminants.In this paper we prepare the nano zero2valent iron particles with a modified method of reduction in liquid phase,in which we add polyvinlpyrrolidone(k30)(which is a kind of macromolecule of dispersant)and ethanol to change the physical property of the surface of the nano zero2valent iron particles in order to meliorate its dispersibility in the water solution.In the experiment of this study,the water solution(or ethanol2water solution)of NaB H4of a certain concentration is added to the water solution(or ethanol2water solution)of FeSO4・7H2O of a certain concentration rapidly with the mechanical agitating,and then lots of iron powder is produced in a short time.The reaction is very fast and it doesn’t need the protection of N2in the process.We adopt the transmission electron microscope(TEM), X2ray diffraction(XRD)and Brunauer Emmett Teller(B ET)to characterize the iron particles.The result of the TEM indicates that the particles are dispersed homogeneously with small average grain diameter of60nm for the water solution and40nm for the ethanol2water solution.The polyvinlpnolidone(k30)added in this experiment plays a very important role in the dispersibility of the nano iron particles by changing the charge distribution of the surface and then generating the effect of electrostatic stabilizing,space steric and electrostatic space steric stabilizing.When ethanol is added in,the size of the nano particles is limited and a limiting f ramework is consequently formed. Perhap s there are many f ree and strong polar hydroxyl groups in the ethanol,and in the water solution these groups chelate to the metallic ions and then cover them tightly.The result of the XRD shows that when the scan angle(2θ) ranges from30°to100°,the corresponding2θwhere the diff raction peak appears are about45°,65°,82°respectively.I f we compare it with the standard PDF card of the iron,we can find the relevant110crystal plane(44.6732°),200 crystal plane(65.0211°)and211crystal plane(82.3326°).With the analysis of Prague equation and electron diff raction,we can conclude that the particles are elementary iron with a high purity,in which there is no iron oxide.At last the result of the B ET shows that the average specific surface area of the particles is47.1m2/g for the water solution and68.41m2/g for the ethanol2water solution,which is higher than that of common iron powder. Results show that this technology is stable,which is proved in our repetitious experiments.K ey w ords: nano,zero2valent iron particles,preparation,liquid phase reduction method 零价铁电负性较大,具有较强的还原性.文献[1,2]的研究均表明:零价铁可以有效地去除水体中氯代脂肪烃类,尤其对氯代烷烃具有较强的降解能力.但是普通铁粉的反应活性比较低,只能部分降解氯代有机化合物,且反应速率较慢,同时由于在铁颗粒表面会形成表面钝化层,导致铁的还原活性随时间下降.因此应用普通零价铁粉来降解有机氯农药、PCDD、PBDE・953・ 第4期高树梅等:改进液相还原法制备纳米零价铁颗粒等方面目前还存在着一些技术瓶颈.纳米铁颗粒因其粒子的直径小,颗粒的比表面积和表面能大,从而具有优越的吸附性能和很高的还原活性.利用纳米颗粒特有的表面效应和小尺寸效应,可以提高零价铁颗粒的反应活性和处理效率,近年来在废水处理方面应用广泛.Lowry et al.[3]研究了纳米铁降解六种PCB的情况; Engelmann et al.[4]则采用双金属系统(Fe/Pd 和Mg/Pd)同时降解PCB和DD T,均取得了很好的降解效果.因此研究纳米铁颗粒的制备方法有着重要的意义,引起了国内外许多学者的广泛关注[5~9].本文采用了一种改进的液相还原法来制备纳米铁颗粒,方法原理简单,制备快捷,实验中添加的聚乙烯吡咯烷酮(PV P K-30)对铁颗粒的分散起到了很好的改善作用,乙醇的添加也使颗粒的粒径降低.1 实验方法与原理1.1 原料与仪器 七水硫酸亚铁(FeSO4・7H2O),硼氢化钠(NaB H4),聚乙烯吡咯烷酮(PV P K-30),丙酮,无水乙醇均为分析纯,蒸馏水,机械搅拌器,天平.1.2 铁颗粒的表面改性及原理 纳米颗粒本身由于表面缺少邻近配位的原子,具有很高的活性,从而使得纳米粒子间存在着有别于常规粒子的自发作用趋势,暂且称这种自发的作用趋势为纳米作用能[10,11],纳米作用能是纳米粒子易团聚的内在因素.纳米铁颗粒更因为其具有磁性,在水溶液中更易发生团聚,从而影响其应用.针对这一现象,我们主要采用通过添加吸附包裹改性剂如高分子分散剂来进行表面物理改性,高分子分散剂含有两类主要成份:一类是活性官能团,它们能通过静电结合使分散剂固定在粒子的表面,另一类是可溶解的大分子链,这些分子在由低到高不同极性的介质中都很适合分散.分散剂分散主要机理是通过分散剂吸附改变粒子的表面电荷分布,产生静电稳定效应(图1),空间位阻作用(图2)和静电空间位阻稳定效应(图3)来达到分散效果[12].图1 静电稳定效应Fig.1 E lectrostalicstabilityeffect图2 空间位阻作用Fig.2 Steric hindranceeffect图3 静电空间位阻稳定效应Fig.3 Etectrostatic sterichindrance effect1.3 纳米铁颗粒的制备 方法一:配制0.01mol/L的FeSO4・7H2O水溶液50mL,然后加入1g聚乙烯吡咯烷酮(PV P K-30),机械搅拌使之充分混合.配制0.03mol/L的NaB H4水溶液.机械搅拌条件下,将50mL NaB H4水溶液迅速添加到50mL FeSO4・7H2O水溶液中,继续搅拌数秒钟,溶液变为黑色时停止搅拌.用磁选法选出,先用蒸馏水充分洗涤3次,然后用丙酮充分洗涤3次,并保存于丙酮中.方法二:在水溶液中加入一定量的乙醇(乙醇:水=1∶9~1∶1),重复上述操作.对于上述工艺,主要有下面几方面的改进(1)采用迅速添加的操作工艺,缩短了反应时间,从而防止了铁颗粒因在水溶液(或乙醇-水混合溶液)中停留时间过长而被氧化.(2)无需氮气保护,操作工艺简单,条件容易控制,操作方便.(3)通过添加高分子分散剂聚乙烯吡咯烷酮(PV P)和乙醇对铁颗粒表面进行改性.1.4 纳米铁颗粒的表征 采用日本J EOL公・063・南京大学学报(自然科学) 第43卷司的J EM -200CX 型透射电子显微镜(TEM )观察粒子的形貌,粒径大小以及团聚情况.采用瑞士A RL 公司的X ’TRA 型X 射线衍射仪(XRD )对产物进行晶粒度分析以及晶体结构分析,采用北京精微高博科技开发中心的J W -04氮吸附比表面测定仪(B ET )来测定颗粒的比表面积.2 结果与讨论2.1 TEM 的表征结果2.1.1 TEM 扫描 透射电子显微镜是目前最常用,最直接的测试手段,利用透射电镜可以较直接的研究纳米颗粒的颗粒分布状态,但可能会有较大的统计误差,同时由于电镜法是对样品局部区域的观测,所以在进行粒度分析时,需要多幅照片的观测,通过分析得到统计的粒度分布[13].因此,本文给出了多幅TEM 扫描图片,以便得出颗粒的粒径分布.廖学红等[14]对纳米银进行粒度表征时也是采用了TEM 进行表征.采用上述两种方法制备的纳米铁颗粒的TEM 扫描图片如图4和图5.图中的比例尺度为100nm ,除图5中的第一张为放大7.5万倍外,其余图片均为放大5万倍.图4 方法一制备的纳米铁颗粒的TEM 图片Fig.4 TEM pictures of nano iron particles prepared by method1图5 方法二制备的纳米铁颗粒的TEM 图片Fig.5 TEM pictures of nano iron particles prepared by method 2・163・ 第4期高树梅等:改进液相还原法制备纳米零价铁颗粒 TEM 的测试结果表明:采用上述两种方法制备的纳米铁颗粒,分散均匀,第一种方法的平均粒径在60nm 左右;添加乙醇后使粒径更小,约40nm 左右.实验过程添加的PV P K -30对颗粒的分散起了很好的改善作用.图中个别位置出现了大粒径颗粒的现象,可能是由于铁颗粒具有磁性而团聚在一起.颗粒呈球状且连成树枝状分布,这是由于磁性纳米粒子受地磁力,小粒子间的静磁力以及表面张力等共同作用的结果[15].其中纳米颗粒的粒径是通过透射电镜照片上30个颗粒的直径平均值求得的[13],d n =∑30i =1D in,其中d n 为颗粒直径平均尺寸,D i 为颗粒直径尺寸.加入乙醇后,可能是由于乙醇中包含大量的自由的强极性羟基基团,在水溶液中这些基团与金属离子之间形成螯合键,紧密包覆在金属离子周围,形成一个有限制形状的有限结构,使合成的纳米粒子的大小被限制,从而达到改性的目的[16].图6 纳米铁颗粒的电子衍射花样图片Fig.6 electron diffraction picture of nano iron p articles张朝平等[17]采用微乳液法,用十二烷基苯磺酸钠(DBS )/异戊醇/正庚烷/H 2O 反应体系,以NaB H 4作还原剂,用FeCl 2・6H 2O 制备纳米级包裹型超细铁粉:采用XRD 、SEM 、TEM 和IR 技术进一步表征了制得的超细铁粉呈均匀球形,平均粒径约120nm ;表面包裹有DBS.与本方法相比,其粒子的粒径较大,且本方法操作简单.采用气相方法来制备纳米铁颗粒,其粒径一般相对较小,但是气相方法对设备要求较高,需高压高温等操作,因此适合大规模工业生产.固相方法相对来说较易引入杂质,纯度不高,一般适合与其它方法联合起来应用.2.1.2 纳米铁颗粒的电子衍射花样 图6为纳米铁颗粒的电子衍射花样扫描图片,仪器的L λ=35.8mm ,L λ称为相机常数,L 为相机长度,λ是电子波的波长.图中可以看出,出现了两个较为明显的衍射环,第一个衍射环所对应的半径R 1≈27mm ,第二个衍射环对应的半径R 2≈30mm.根据标定电子衍射花样常用的公式D d =L λ[17]知:d 1=LλR 1=35.8mm !27mm =2.106d 2=LλR 2=35.8mm !30mm=1.193 对照零价铁的标准PDF 卡片发现此两晶面刚好对应相应的110晶面(d =2.0268),211晶面(d =1.1702).表1为零价铁的标准PDF 卡片的相关参数.表1 零价铁的标准PDF 卡片的相关参数T able 1 Stand ard PDF cards of zero 2valent iron 衍射晶面晶面间距(!)衍射角(°)相对衍射强度110 2.026844.6732100200 1.433265.021120211 1.170282.332630220 1.013498.6451103100.9064116.3849122220.8275137.13606图7 纳米铁颗粒的XR D 表征结果Fig.7 XR D diagram of nano iron particles・263・南京大学学报(自然科学) 第43卷2.2 XR D的表征结果 图7为纳米铁颗粒的X射线衍射图,扫描的2θ角度为30°~100°,入射波的波长为1.5418. XRD的测试结果表明:在扫描衍射角度(2θ)为30°~100°时,出现衍射峰时对应的2θ分别为44.59°,64.03°,81.84°,对照铁的标准PDF卡片发现,刚好对应相应的110晶面衍射(44.6732°),200晶面衍射(65.0211°),211晶面衍射(82.3326°).表明颗粒为单质铁,而没有出现氧化铁物质.根据布拉格方程2d sinθ=nλ[19],d为相邻平行晶面的晶面间距,θ为入射角,λ为入射波的波长,n为衍射级数,在此取1,可以得出相应的晶面间距d1=λ2sinθ1=1.5418!2sin22.3=2.0316!d2=λ2sinθ2=1.5418!2sin32°=1.4547!d3=λ2sinθ3=1.5417!2sin40.9°=1.1821! 对照表1发现:刚好对应相应的110晶面衍射(d=2.0268!),200晶面衍射(d=1.4332 !),211晶面衍射(d=1.1702!).从上述几种表征及计算的结果均看出:该方法所得颗粒为单质铁,过程中没有出现氧化铁杂质,纯度高.其中电子衍射花样中没有出现200晶面衍射环的原因可能是由于衍射强度较小的原因,从图6中也可以看出200晶面的衍射强度远小于110晶面衍射和211晶面衍射.2.3 BET的表征结果 采用氮吸附表面测定仪(B ET)的测定结果表明:采用方法一制备的纳米铁颗粒的比表面积为47.1m2/g;采用方法二制备的纳米铁颗粒的比表面积为68.41 m2/g,此结果也刚好与上述TEM的表征结果相对应,即粒径小,比表面积大.也再次证明了方法的稳定性.目前,国内外的许多实验室都在做相关的研究.Sung et al.[20]采用液相还原法制备了纳米铁颗粒用来降解除草剂,经B ET分析,制备的零价铁颗粒比表面积为32m2/g,粒径分布为1~200nm,平均粒径为50nm.Liao et al.[14]采用液相还原法制备了钯/铁双金属纳米颗粒用于降解氯代烃类污染物,他们制得的纳米铁颗粒粒径在10~100nm,比表面积为35 m2/g.SU SHIL RAJ KAN EL等[21]采用该方法制备纳米级铁颗粒用来去除地表水中的三价砷,A FM分析,颗粒粒径在1~120nm.本文所采用的方法与上述实验室的结果相比较,颗粒的比表面积有明显的提高.3 结 论(1)该方法无需氮气保护,对设备的要求相对较低,同时方法原理也相对简单;该方法最大的优点是反应迅速,制备快捷,可以在短时间内产生大量铁粉.(2)溶液中添加的高分子分散剂聚乙烯吡咯烷酮(PV P)对纳米铁颗粒的分布起到了很好的分散作用.平均粒径在60nm左右,比表面积为47.1m2/g;添加的无水乙醇使颗粒的粒径减小,比表面积增大,平均粒径在40nm左右,比表面积为68.41m2/g,远高于普通铁粉.(3)多次试验的结果表明,该方法非常稳定.R eferences[1] Quan X,Liu H J,Yang F L,et al.Dechlorina2tion of three polychlorinated hydrocarbons in wa2ter using bimetallic systems.China EnviromentalScience,1998,18(4):333~336.(全 燮,刘会娟,杨凤林等.二元金属体系对水中多氯有机物的催化还原脱氯特性.中国环境科学,1998,18(4):333~336.)[2] He X J,Liu F,Huang Y Y,et al.Degradationof volatile chlorinated aliphatics by zero2valentIron.Environmental Science,2003,24(1):139~142.(何小娟,刘 菲,黄园英等.利用零价铁去除挥发性氯代脂肪烃的试验.环境科学,2003,24(1):139~142.)[3] Lowry G V,Johnson K M.Congener2specific de2chlorination of dissolved PCBs by microscale andnanoscale zerovalent Iron in a water/methanolSolution.Environ Sci Technol,2004,38(19):5208~5216.[4] Engelmann M D,Hutcheson R,Kristy H.Sim2ultaneous determination of total polychlorinatedbiphenyl and dichlorodiphenyltrichloroethane・363・ 第4期高树梅等:改进液相还原法制备纳米零价铁颗粒(DD T)by dechlorination with Fe/Pd and Mg/Pdbimetallic particles and flame ionization detectiongas chromatography.Microchemical Journal,2003,74:19~25.[5] G edanken ing sonochemistry for the fabri2cation of nanomaterial ultrasonics.Sonochemis2try,2004,11:47~55.[6] Khomutov G B,Bykov I V,Gainutdinov R V,etal.Two2dimensional photochemical synthesis ofplate2like nanoparticles.Physicochemical and En2gineering Aspects,2002,198~200:347~358..[7] Liu J B,Dong W,Zhan P.Synthesis of bimetal2lic nanoshells by an improved electroless platingngmuir,2005,21:1683~1686. [8] Grirrane A,Pastor A,Mealli C.Synthesis,str2ucture,magnetic and electrochemical propertiesof an oxydiacetate iron(II)complex.NorganicaChimica Acta,2004,57:4215~4219.[9] Cheng W H,Wu K C,Lo M Y.Recent advancesin nano precious metal catalyst research at UnionChemical Laboratories.Catalysis Today,2004,97:145~151.[10] Xing H L,Xu G C,Li A C,et al.Develop2ment in dispersion of nanaoparticles and prepa2ration of organic2inorganic nonocomposites,Material Review,2001,15(9):62~64.(邢宏龙,徐国财,李爱元等.纳米粉体的分散及纳米复合材料的成型技术.材料导报,2001,15(9):62~64). [11] Xu G C,Ma J J,Xing H L.Dispersion of nan2opaerticles and organic nonocomposites.Bulle2tin of National Natural Science Foundation ofChina,2001,15(2):109~112.(徐国财,马家举,邢宏龙.纳米粒子的分散及其有机复合材料的复合技术.中国科学基金,2001,15(2):109~112). [12] Wan D L,Wang Y,Bai Q D.Dispersion andmodified technique of nanoparticles power inwater medium.Science and Technology ofOverseas Building Materials,2005,26(1):25~28.(万德立,王 勇.白清东.纳米粉体在水性介质中的分散及改性技术.国外建材科技,2005,26(1):25~28.)[13] Zhu Y F.Characterization and measuring andtesting technique of Nanaphase materials.Bei2jing:Chemical Industry Publishing,2006,1~95.(朱永法.纳米材料的表征及测试技术.北京:化学工业出版社,2006,1~95.)[14] Liao X H,Zhu J J,Qiu X F,et al.Preparationof like2spheres and dendrites silver nanoparticlesby sonodlectrochemical method.Journal of Nan2jing University(Natural Sciences),2002,38(1):119~123.(廖学红,朱俊杰,邱晓峰等.类球形和树枝状纳米银的超声电化学制备.南京大学学报(自然科学),2002,38(1):119~123).[15] Wang C Y,Chen Z Y,Cheng B,et al.Thepreparation,surface,modification,and charac2terization of metallicα2Fe nanoparticles.ActaPhysics Sinica,1999,12(6):670~674.(王翠英,陈祖耀,程 彬等.金属铁纳米粒子的液相制备.表面修饰及其结构表征.化学物理学报,1999,12(6):670~674).[16] Jiu J T,Li L P,Ge Y,et al.The Preparationof MgO nanoparticles protected by polymer.Chinese Journal of Inorganic Chemistry,2001,17(3):361~365.(酒金婷,李立平,葛 钥等.用高分子保护的纳米MgO的合成.无机化学学报,2001,17(3):361~365).[17] Zhang C P,Deng W,Hu Z C,et al.Prepara2tion of ultrafine Fe particles by microemulsionmethod.Chinese Journal of Applied Chemistry,2000,17(3):248~252.(张朝平,邓 伟,胡宗超等.微乳液法制备超细包裹型铁粉.应用化学,2000,17(3):248~252).[18] Huang H Z.Nanaphase materials analyze.Bei2jing:Chemical Industry Publishing,2003,260~280.(黄惠忠.纳米材料分析.北京:化学工业出版社,2003,260~280).[19] Xu B D.Nanaphase materials and application tech2nology.Beijing:Chemical Industry Publishing,2004,123~124.(许并社.纳米材料及应用技术.北京:化学工业出版社,2004,123~124).[20] Sung H J,Andrew J,Feitz A,et al,Oxidativedegradation of the carbothioate herbicide,moli2nate,using nanoscale zero2valent Iron.Environ2mental Science and Technology.2004,38,2242~2247.[21] Lien H L,Zhang W X.Nanoscale iron particlesfor complete reduction of chlorinated ethenes.Colloids and Surfaces A:Physicochemical andEngineering Aspects,2001,191:97~105.・463・南京大学学报(自然科学) 第43卷。
液相法制备纳米颗粒的机制液相法是在液体状态下通过化学反应制备纳米材料方法的总称,又称为湿化学法或溶液法。
纳米材料的液相制备方法分为:沉淀法、溶胶-凝胶(Sol-Gel)法、水热法、化学还原法、化学热分解法、微乳液法、声化学法、电化学法和水中放电法等9种。
用液相化学法合成纳米颗粒能够较好地控制颗粒大小、形状和粒径分布。
为了充分利用化学法的优点,需要充分了解这种方法制备纳米颗粒的形成机制,这涉及到:晶体化学、热力学、相平衡以及反应动力学的基本原理。
从液相中生成固相颗粒,要经过成核、生长、凝结、团聚等过程。
1 液相中生成固相颗粒的机理1.1 成核为了从液相中析出大小均匀一致的固相颗粒,必须使成核和长大这两个过程分开,以便使已成核的晶核同步地长大,并在生长过程中不再有新核形成。
在纳米颗粒形成的最初阶段,都需要有新相的核心形成。
新相的形核过程可以被分为两种类型,即自发形核与非自发形核过程。
所谓自发形核指的是整个形核过程完全是在相变自由能的推过下进行的,而非自发形核则指的是除了有相变自由能作推动力之外,还有其他的因素起到了帮助新相核心生成的作用。
图1 析出固体时液相中溶质浓度随时间的变化情况如图1所示,在整个成核和生长过程中液相内与析出物相应的物质的量浓度是变化的。
在阶段Ⅰ浓度尚未达到成核所要求的最低过饱和浓度*min c ,因此无晶核形成。
当液相中溶液浓度超过*min c 后即进入成核阶段Ⅱ。
作为自发形核的例子,我们考虑一个从过饱和溶液中析出一个球形的固相核心的过程。
设新相核心的半径为r ,因而形成一个新相核心时体自由能将变化343C r G π∆,其中C G ∆为从溶液中析出单位体积晶核时伴随的自由能变化。
0ln C Tc G V c κ∆=- (1-1)上式还可以写成:()ln 1C TG s V κ∆=-+ (1-2)其中,c 为过饱和溶液的浓度;0c 为饱和溶液的浓度;V 晶体中单个分子所占的体积;()00s c c c =-是液相的过饱和度。
当过饱和度为零时,0C G ∆=,这时将没有新相的核心可以形成,或者已经形成的新相核心不能获得长大。
当液相存在过饱和现象时,0C G ∆<,它就是新相形核的驱动力。
在新的核心形成的同时,还将伴随有新的固-液相界面的生成,它导致相应界面能的增加,其数值为24r πγ,其中γ为单位面积的界面能。
综合上面两项能量之后,我们得到系统的自由能变化为:32443C G r G r ππγ∆=∆+ (1-3)将上式对r 微分,求出使得自由能G ∆为零的条件为: *2C r G γ=-∆ (1-4)它是能够平衡存在的最小的固相核心半径,又称为临界核心半径。
当*r r <时,在热涨落过程中形成的这个新相核心将处于不稳定状态,它将可能再次消失。
相反,当*r r >时,新相的核心将处于可以继续稳定生长的状态,并且生长过程将使得自由能下降。
将式(1-4)代入(1-3)后,可以求出形成临界核心时系统的自由能变化:3*2163C G G πγ∆=∆ (1-5)图2 新相形核过程的自由能变化随核心半径的变化趋势图2中画出了形核自由能变化随新相核心半径的变化曲线。
我们看到,形成临界核心的临界自由能变化*G ∆实际上就相当于形核过程的能垒。
热激活过程提供的能量起伏将使得某些原子集团具备了*G ∆大小的自由能涨落,从而导致了新相核心的形成。
液相中均匀成核的核生长速率可用式(1-6)表示:0exp exp C D G G J J T T κκ∆∆⎛⎫⎛⎫= ⎪ ⎪⎝⎭⎝⎭ (1-6)式中,0J 为分子的跃迁频率;D G ∆为晶核在液相中的扩散活化自由能;C G ∆为从溶液中析出晶核时伴随的自由能变化;κ为玻耳兹曼常数;T 为热力学温度。
核的生长速率随的变化而很快地变化。
非均匀成核时,在相界表面上(如外来质点、容器壁以及原有晶体表面上)形成晶核,称非均匀成核,临界核生成的自由能变化为*C G ∆。
可用式(1-7)式表示:()()2*2cos 1cos 4C C G G θθ⎡⎤+-∆=∆⎢⎥⎢⎥⎣⎦(1-7) 式中,θ是液体和固体形成的接触角,由于()()22cos 1cos 41θθ+-<,所以*CG ∆比均匀成核的C G ∆要小。
非均匀核的成核速率可用式(1-8)表示:()()()22231222cos 1cos 2162exp 34CV CV V V V V J n P m T G T G θθσπσππκκ-⎡⎤+-⎛⎫=-⨯⎢⎥ ⎪∆∆⎢⎥⎝⎭⎣⎦(1-8) 式中,V n 为蒸汽或液相的密度;P 为压强;V G ∆为亚稳相中单相原子或分子转变为稳定相中单个原子或分子所引起的自由能的变化;m 为分子的质量;CV σ为比表面能。
从式(1-8)可以看出,核生长速率对V G ∆值是非常敏感的,不均匀核生成比均匀核的生成容易。
式(1-8)对液相或气相中的非均匀成核皆适用。
阶段Ⅲ是生长阶段,晶体的生长是在生成的晶核上吸附原子或分子而使其长大。
1.2 生长1.2.1 四种界面模型(1)完整光滑界面结构模型(Kossel 模型)1927年,Kossel 提出了完整光滑界面结构模型。
模型认为,晶体是理想完整的,生长界面在原子层次上没有凹凸不平,固相和液相之间是突变的。
晶体生长时,光滑面上首先发生二维成核,一旦晶核形成,生长界面上就会出现台阶,在台阶上必然存在三面角位置(称为扭折)。
这些位置束缚能最大,最容易吸附原子。
对应于完整光滑界面的晶体生长包括以下过程:①原子从稀薄环境相向扭折处作三维扩散;②吸附原于在生长界面上向扭折处作二维扩散;③扭折的延伸,台阶的扩展,界面逐渐铺满原于,进而转变成为新的完整光滑界面。
晶面的生长速率取决于界面上扭折密度以及扭折处吸附原子的能力。
Kossel 模型选用了简单的立方结构晶体,仅考虑单个原子在生长界面上的叠合,因此是一种非常简单化的理想界面,与实际晶体生长有很大的距离。
(2)非完整光滑界面结构模型1949年,Frank提出了非完整光滑面界面结构模型,它是Kossel-Stranski 理论的发展。
模型认为晶体生长界面不是理想完整的,界面上存在一定数量的位错。
如果一个纯螺旋型位错和光滑的奇异面相交,在晶面上就会产生一个永不消失的台阶源。
在晶体生长过程中,台阶将逐渐变成螺旋状,使晶面不断向前推移。
后来,Burton、Cabrera和Frank进一步发展了该模型,形成了较为完整的Burton-Cabrera-Frank晶体生长理论(简称为BCF理论)。
(3)粗糙化界面结构模型1958年,Jackson提出了粗糙化界面模型,其基础是考察恒温恒压条件下,生长界面层中流体相原子转变为晶相原子所引起的自由能变化。
模型假设晶体生长界面为单原子层,生长界面中包含的晶相和流体相原子都位于品格位置上;吸附原子进入到生长界面是随机的,液体是连续的流体,流体原子之间、晶相原子与流体原子之间没有相互作用,表面键能只考虑吸附原子之间最近邻的相互作用。
根据统计热力学近似计算,可以判断固/液界面的平衡结构性质。
判断其是光滑界面(界面层全部为晶相原子)还是粗糙界面(界面层晶相原子和流体相原子各占一半)。
一般来讲,如果晶体相变熵小于2,生长界面为粗糙界面;如果晶体相变熵大于4,生长界面为光滑界面;如果晶体相变熵在2—4之间,生长界面是光滑还是粗糙界面,不仅取决于相变熵,而且还取决于生长界面的取向因素等。
(4)弥散界面结构模型1966年,Temkin提出了弥散界面结构模型(又称为多层界面模型)。
模型考虑正方晶系晶体,认为生长界面由多个原子层组成,界面晶格位置由晶相原子和流体相原子所占据,在整个生长过程中,“晶相块”仅能在晶相块上堆积,仅考虑“晶相块”和“流体相块”之间、“晶相块”之间和“流体相块”之间最近邻的相互作用。
弥散界面结构模型具有以下特点:①和Jackson单原子层模型相比,模型没有限制原子层数,因此更“贴近”实际晶体生长情况;②使用平衡热力学基本原理,处理较为简单;③模型不仅适用于固/液生长界面,也适用于固/气或固熔体生长界面。
弥散界面结构模型是目前应用最广的晶体生长界面模型。
1.2.2 生长机理研究(1)二维成核生长模型当晶体在气相或溶液中生长时,若生长界面为原子级完整光滑界面时,晶体生长遵循二维成核生长机制。
原子或分子被吸附到生长界面后,通过扩散聚集而形成二维晶核。
二维晶核一旦出现,体系就增加了棱边能。
此棱边能效应与三维晶核中界面能效应完全类似,构成了二维晶核的热力学势垒。
因此,只有当尺寸达到临界大小时,二维晶核才能自发生长。
以n t 表示连续两次二维成核时间间隔,以s t 表示1个二维临界晶核的台阶“扫过”整个生长界面所需要的时间,根据n t 和s t 的关系,可把二维成核生长分为两种类型“。
一是单二维成核生长,即当n s t t ≥时,在新的二维成核再次形成以前,有足够时间让该晶核的台阶“扫过”整个生长界面;二是多二维成核生长,即当n s t t ≤时,单二维晶核的台阶扫过整个生长界面所需要的时间远远超过连续两次成核的时间间隔,即生长界面每增长1个原子层,需要2个以上的二维品核。
根据原于(分子)成核方式,二维成核又分为表而扩散二维成核(Surface Diffusion two-Dimensional Nucleation)和直接在扭折处叠合二维成核(Direct Integration two-Dimensional Nucleation)。
(2)螺位错生长模型若生长界面上有螺位错露头点,晶体生长机制与二维成核机制不同。
此时,晶体生长起源于生长界面上螺旋位错露头点的台阶,在生长过程中台阶永不消失,螺旋位错露头点提供了一个连续起作用的台阶源,生长界面为一连续的螺旋面。
假设台阶高度为h ,相邻螺旋之间的距离为1919c r kTs λγ==Ω,形成螺旋状生长丘的斜度p h λ=。
如果台阶运动速率为ν,晶体生长的法向生长速率为:n R h p υλν==,与二维成核生长相似,螺旋位错生长也分为表面扩散和直接在扭折处叠合两种形式。
(3)体扩散控制生长模型将在生长界面上吸附结晶的粒子或粒子团称为生长基元,将生长基元在结晶相(即已形成的晶体)内的扩散速度称为体扩散速度。
如果生长基元的体扩散速度小于其在生长界面上的扩散速度,或者小于在生长界面扭折处叠合(结晶)的速度时,晶体生长的速度就由体扩散速度决定,相应的晶体生长称为体扩散控制的生长。
1.2.3 界面生长晶体界面生长,是生长基元不断从流体相通过界面进入晶格位置的过程,也是晶体和流体界面不断向流体中推移的过程,即晶体界面生长的过程是气相或者液相的原子或分子扩散到晶体表面附着并进入晶格。