无源植入性医疗器械货架寿命申报资料指导原则
- 格式:doc
- 大小:41.50 KB
- 文档页数:11


5、医用外科口罩研究资料XXXXXXX有限公司2020年4月5.1医用外科口罩产品性能研究5.1.1医用外科口罩产品性能及技术指标2. 性能指标2.1 外观口罩外观应整洁、形状完好,表面不得有破损、污渍。
2.2结构与尺寸口罩佩戴好后,应能罩住佩戴者的口、鼻至下颌。
应符合设计的尺寸,最大偏差应不超过±5%。
2.3 鼻夹2.3.1口罩上应配有鼻夹,鼻夹由可塑性材料制成。
2.3.2鼻夹长度应不小于8.0cm。
2.4口罩带2.4.1口罩带应戴取方便。
2.4.2每根口罩带与口罩体连接点处的断裂强度应不小于10N。
2.5合成血液穿透2ml合成血液以16kPa(120mmHg)压力喷向口罩外侧面后,口罩内侧面不应出现渗透。
2.6过滤效率2.6.1细菌过滤效率(BFE)口罩的细菌过滤效率应不小于95%。
2.6.2颗粒过滤效率(PFE)口罩对非油性颗粒的过滤效率应不小于30%。
2.7压力差口罩两侧面进行气体交换的压力差ΔP应不大于49Pa/cm2。
2.8阻燃性能口罩材料应采用不易燃材料;口罩离开火焰后燃烧不大于5s。
2.9无菌2.9.1环氧乙烷灭菌2.10环氧乙烷残留口罩如经环氧乙烷灭菌或消毒,其环氧乙烷残留量应不超过10μg/g。
5.1.2 产品性能及技术指标确定的依据5.1.2.1产品性能和技术要求的确定依据的说明医用外科口罩产品性能和技术要求的确定依据,我公司依据国家标准和行业标准中相关要求,在研制过程中经摸索试验并参考了国内同类产品性能指标、技术要求,并结合多家医疗机构临床专家的临床经验基础上确定而成。
按照技术成熟、经济合理、安全可靠和符合临床要求的基本原则。
根据医用外科口罩产品本身特点,采用规定程序批准的工艺制作,参照国家标准和行业标准中相关要求,产品技术要求规定了产品的型号规格、性能要求和检测方法。
结合预期用途、产品功能、确定了产品性能要求的内容为外观;结构与尺寸;鼻夹;口罩带;细菌过滤效率(BFE);颗粒过滤效率(PFE);压力差;阻燃性能;合成血液穿透;无菌;环氧乙烷残留。

金属接骨板内固定系统产品注册技术审查指导原则一、前言金属接骨板内固定系统作为骨科植入性医疗器械,是治疗骨折的主要手段之一,其安全性和有效性直接影响骨折的治疗效果。
本指导原则旨在为申请人/生产企业进行金属接骨板内固定系统的注册申报提供技术指导,同时也为食品药品监督管理部门对注册申报资料的审评提供技术参考。
本指导原则系对金属接骨板内固定系统注册申报资料的一般要求,申请人/生产企业应依据具体产品的特性对注册申报资料的内容进行充实和细化,并依据具体产品的特性确定其中的具体内容是否适用。
本指导原则是对申请人/生产企业和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规和标准的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
二、适用范围本指导原则涵盖的金属接骨板内固定系统适用于四肢骨(包括上肢带骨:锁骨、肩胛骨,自由上肢骨:肱骨、桡骨、尺骨、腕骨、掌骨、指骨,下肢带骨:髋骨、坐骨、耻骨;自由下肢骨:股骨、髌骨、胫骨、腓骨、跗骨、跖骨、趾骨)骨折内固定;由非锁定金属接骨板、非锁定金属接骨螺钉、锁定金属接骨板、锁定金属接骨螺钉和空心螺钉组成;由外科植入物用金属材料制成,包括纯钛、Ti6Al4V钛合金、TC4钛合金、TC4ELI钛合金、Ti6Al7Nb钛合金、00Cr18Ni14Mo3不锈钢、00Cr18Ni15Mo3N不锈钢、高氮不锈钢、锻造钴铬钼合金。
本指导原则不适用特殊设计及创新设计的产品。
三、基本要求(一)注册单元的划分金属接骨板内固定系统可按照实现某种临床预期用途的产品组合划分注册单元,亦可以组件作为注册单元进行申报:1.若按照实现某种临床预期用途的产品组合进行申报,金属接骨板内固定系统包括如下注册单元:(1)直型非锁定金属接骨板系统:由直型非锁定金属接骨板、非锁定金属接骨螺钉组成。

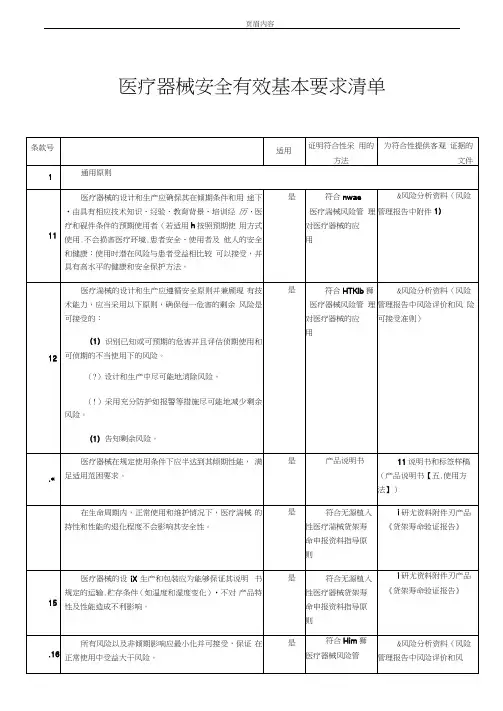
3.医疗器械安全有效基本要求清单医疗器械安全有效基本要求清单条款号要求适用证明符合性采用的方法为符合性提供客观证据的文件A 通用原则A1 医疗器械的设计和生产应确保其在预期条件和用途下,由具有相应技术知识、经验、教育背景、培训经历、医疗和硬件条件的预期使用者(若适用),按照预期使用方式使用,不会损害医疗环境、患者安全、使用者及他人的安全和健康;使用时潜在风险与患者受益相比较可以接受,并具有高水平的健康和安全保护方法。
是符合YY/T0316-2008 医疗器械风险管理对医疗器械的应用8.风险分析资料(风险管理报告中附件1)A2 医疗器械的设计和生产应遵循安全原则并兼顾现有技术能力,应当采用以下原则,确保每一危害的剩余风险是可接受的:(1)识别已知或可预期的危害并且评估预期使用和可预期的不当使用下的风险。
(2)设计和生产中尽可能地消除风险。
(3)采用充分防护如报警等措施尽可能地减少剩余风险。
(4)告知剩余风险。
是符合YY/T0316-2008 医疗器械风险管理对医疗器械的应用8.风险分析资料(风险管理报告中风险评价和风险可接受准则)A3 医疗器械在规定使用条件下应当达到其预期性能,满足适用范围要求。
是产品说明书11.说明书和标签样稿(产品说明书【五、使用方法】)A4 在生命周期内,正常使用和维护情况下,医疗器械的特性和性能的退化程度不会影响其安全性。
是符合无源植入性医疗器械货架寿命申报资料指导原则5.研究资料附件5-7产品《货架寿命验证报告》A5 医疗器械的设计、生产和包装应当能够保证其说明书规定的运输、贮存条件(如温度和湿度变化),不对产品特性及性能造成不利影响。
是符合无源植入性医疗器械货架寿命申报资料指导原则5.研究资料附件5-7产品《货架寿命验证报告》A6 所有风险以及非预期影响应最小化并可接受,保证在正常使用中受益大于风险。
是符合YY/T0316-2008 医疗器械风险管理对医疗器械的应用8.风险分析资料(风险管理报告中风险评价和风险可接受准则)B 医疗器械安全性能基本原则B1 化学、物理和生物学性质B2.7 若医疗器械可以以无菌与非无菌两种状态上市,则产品的包装或标签应当加以区别。

附件2髋关节假体系统注册技术审查指导原则本指导原则旨在为申请人进行髋关节假体系统产品的注册申报提供技术指导,同时也为食品药品监督管理部门对注册申报资料的审评提供技术参考。
本指导原则系对髋关节假体系统产品注册申报资料的一般要求,申请人应依据具体产品的特性对注册申报资料的内容进行充实和细化,并依据具体产品的特性确定其中的具体内容是否适用。
本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规和标准的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
一、适用范围本指导原则涉及的产品适用于人工髋关节假体置换,包括髋臼部件(髋臼外杯、紧固螺钉、髋臼内衬、整体式髋臼)、股骨部件[股骨头(包含符合行标要求的单极头及双极头)、股骨颈、股骨柄]、中置器、骨水泥远端塞等,由YY 0118《关节置换植入物髋关节假体》标准(注:本指导原则中所列标准适用最新版本,下同)中已认可的金属材料、高分子材料或陶瓷材料制成。
本指导原则不包含表面置换型髋关节假体、特殊设计或个性化定制的髋关节假体。
二、注册单元划分髋关节假体系统可按照系统划分注册单元,亦可以组件为注册单元进行申报:(一)若按照系统进行申报,根据产品的固定方式、固定原理、适应证,将产品划分如下注册单元:1.骨水泥型髋关节假体2.非骨水泥型髋关节假体3.混合型髋关节假体4.短柄髋关节假体(通常CT值≤120mm,保留皮质环,近端固定)(二)若以同一系统内单一组件或多个组件为注册单元进行申报,须明确与该产品配合的组件名称。
(三)材质(如有涂层时,包括涂层)不同的同类组件应划分为不同的注册单元,作为单一整体组配或组合使用的组件可以按同一注册单元申报。

髋关节假体系统注册技术审查指导原则(征求意见稿)一、前言髋关节假体系统作为骨科植入性医疗器械,在治疗损伤或患病的髋关节时发挥重要作用,其安全性及有效性直接影响治疗的效果。
本指导原则旨在为申请人进行髋关节假体系统产品的注册申报提供技术指导,同时也为食品药品监督管理部门对注册申报资料的审评提供技术参考。
本指导原则系对髋关节假体系统产品注册申报资料的一般要求,申请人应依据具体产品的特性对注册申报资料的内容进行充实和细化,并依据具体产品的特性确定其中的具体内容是否适用。
本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行。
如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规和标准的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
二、指导原则适用范围本指导原则涉及的产品适用于人工髋关节假体置换,包括髋臼部件(髋臼外杯、紧固螺钉、髋臼内衬、整体式髋臼)、股骨部件(股骨头(含双极头与行标一致)、股骨颈、股骨柄)、中置器、骨水泥远端塞等,由YY0118《关节置换植入物髋关节假体》标准中已认可的金属材料、高分子材料或陶瓷材料制成。
本指导原则不包含表面置换型髋关节假体、特殊设计或个性化定制的髋关节假体。
三、注册单元的划分髋关节假体系统可按照系统划分注册单元,亦可以组件为注册单元进行申报:(一)若按照系统进行申报,根据产品的固定方式、固定原理、适应症,将产品划分如下注册单元:1、骨水泥型髋关节假体、2、非骨水泥型髋关节假体、3、混合型髋关节假体、4、短柄髋关节假体(通常CT值≤120mm)。
(二)若以同一系统内单一组件或多个组件为注册单元进行申报,须明确与该产品配合的组件名称。
(三)材质不同的同类组件应划分为不同的注册单元,做为单一整体组配或组合使用的组件可以按同一注册单元申报。
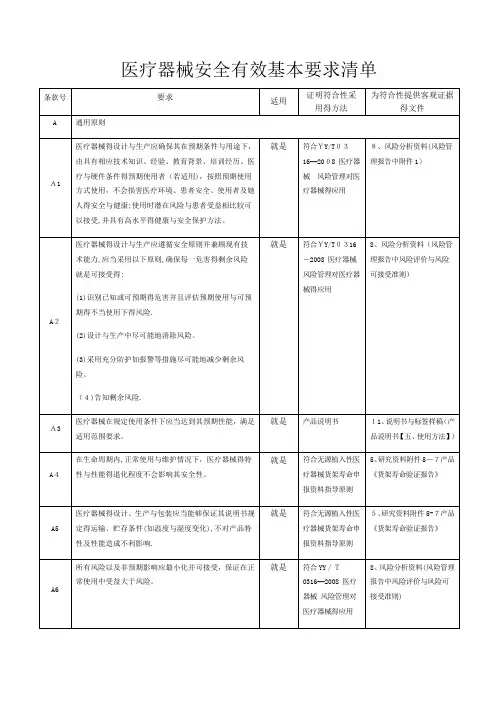


序号名称文件号地址1 白蛋白测定试剂(盒)注册技术审查指导原则2016年第29号/WS01/CL0087/145956.html2 糖化血红蛋白测定试剂盒(酶法)注册技术审查指导原则3 乳酸脱氢酶测定试剂盒注册技术审查指导原则4 促甲状腺素检测试剂注册技术审查指导原则5 甘油三酯测定试剂盒注册技术审查指导原则6 唾液酸检测试剂盒(酶法)注册技术审查指导原则7 β2-微球蛋白检测试剂盒(胶乳增强免疫比浊法)注册技术审查指导原则8 碱性磷酸酶测定试剂盒注册技术审查指导原则(2016年修订版)2016年第28号/WS01/CL0087/145955.html9 人绒毛膜促性腺激素检测试剂(胶体金免疫层析法)注册技术审查指导原则(2016修订版)10 C反应蛋白测定试剂盒注册技术审查指导原则(2016年修订版)11 大便隐血(FOB)检测试剂盒(胶体金免疫层析法)注册技术审查指导原则(2016年修订版)12 缺血修饰白蛋白测定试剂盒注册技术审查指导原则(2016年修订版)13 肌酸激酶测定试剂(盒)注册技术审查指导原则(2016年修订版)14 医学图像存储传输软件(PACS)注册技术审查指导原则2016年第27号/WS01/CL0087/145954.html15 正压通气治疗机注册技术审查指导原则16 大型蒸汽灭菌器注册技术审查指导原则17 腹膜透析机注册技术审查指导原则18 医用内窥镜冷光源注册技术审查指导原则19 振动叩击排痰机注册技术审查指导原则20 磁疗产品注册技术审查指导原则(2016年修订版)2016年第22号/WS01/CL0087/144721.html21 电子血压计(示波法)注册技术审查指导原则(2016年修订版)22 X射线诊断设备(第二类)注册技术审查指导原则(2016年修订版)23 尿液分析仪注册技术审查指导原则(2016年修订版)24 半自动化学发光免疫分析仪注册技术审查指导原则(2016年修订版)25 凝血分析仪注册技术审查指导原则(2016年修订版)26 生化分析仪注册技术审查指导原则(2016年修订版)27 血糖仪注册技术审查指导原则(2016年修订版)28 血液透析用制水设备注册技术审查指导原则(2016年修订版)29 牙科综合治疗机注册技术审查指导原则(2016年修订版)30 医用雾化器注册技术审查指导原则(2016年修订版)31 助听器注册技术审查指导原则(2016年修订版)32 自动尿液有形成分分析仪注册技术审查指导原则(2016年修33 高频手术设备注册技术审查指导原则2016年第21号/WS01/CL0087/144620.html34 医用X射线诊断设备(第三类)注册技术审查指导原则(2016年修订版)35 植入式心脏起搏器注册技术审查指导原则(2016年修订版)36 脉搏血氧仪设备临床评价技术指导原则37 治疗呼吸机注册技术审查指导原则38 强脉冲光治疗仪注册技术审查指导原则39 可吸收止血产品注册技术审查指导原则2016年第7号/WS01/CL0087/143162.html40 腹腔、盆腔外科手术用可吸收防粘连产品注册技术审查指导原则41 透明质酸钠类面部注射填充材料注册技术审查指导原则42 一次性使用膜式氧合器注册技术审查指导原则2016年第6号/WS01/CL0087/143160.html43 α-氰基丙烯酸酯类医用粘合剂注册技术审查指导原则44 丙型肝炎病毒核糖核酸测定试剂技术审查指导原则2015年第93号/WS01/CL1107/136485.html45 过敏原特异性IgE抗体检测试剂技术审查指导原则46 人乳头瘤病毒(HPV)核酸检测及基因分型试剂技术审查指导原则47 全自动化学发光免疫分析仪技术审查指导原则48 医疗器械软件注册技术审查指导原则2015年第50号/CL0056/3810.html49 乙型肝炎病毒基因分型检测试剂技术审查指导原则2015年第32号/CL0057/3808.html50 影像型超声诊断设备新技术注册技术审查指导原则2015年第33号/CL0056/3809.html51 雌激素受体、孕激素受体抗体试剂及检测试剂盒技术审查指导原则2015第11号/CL0057/3553.html52 植入式心脏电极导线产品注册技术审查指导原则 2014第10号 /CL0056/2965.html53 医用控温毯产品注册技术审查指导原则 2014年第7号 /CL0027/2969.html54 电动洗胃机产品注册技术审查指导原则 55 医用电子体温计产品注册技术审查指导原则 56 脉搏血氧仪产品注册技术审查指导原则 57 牙科手机产品注册技术审查指导原则58 C 反应蛋白定量检测试剂盒产品注册技术审查指导原则 59 缺血修饰白蛋白测定试剂产品注册技术审查指导原则 60 肌酸激酶测定试剂盒产品注册技术审查指导原则 61 碱性磷酸酶检测试剂盒产品注册技术审查指导原则 62 医用口罩产品注册技术审查指导原则 63 一次性使用引流管产品注册技术审查指导原则64 护脐带产品注册技术审查指导原则65 一次性医用喉罩产品注册技术审查指导原则66 一次性使用皮肤缝合器产品注册技术审查指导原则67 牙科树脂类充填材料产品注册技术审查指导原则2014年第6号/CL0055/2971.html68 一次性使用避光输液器产品注册技术审查指导原则69 一次性使用血液分离器具产品注册技术审查指导原则70 血液透析浓缩物产品注册技术审查指导原则71 金属接骨板内固定系统产品注册技术审查指导原则72 心脏射频消融导管产品注册技术审查指导原则2014年第5号/CL0056/2970.html73 软性亲水接触镜说明书编写指导原则2014年第3号/CL0055/2972.html74 硬性角膜接触镜说明书编写指导原则75 医用磁共振成像系统注册技术审查指导原则2014年第2号/CL0056/2973.html76 弓形虫、风疹病毒、巨细胞病毒、单纯疱疹病毒抗体及G型免疫球蛋白抗体亲合力检测试剂技术审查指导原则77 肿瘤个体化治疗相关基因突变检测试剂技术审查指导原则78 药物滥用检测试剂技术审查指导原则79 红外线治疗设备产品注册技术审查指导原则2013年第8号/CL0027/2974.html80 中频电疗产品注册技术审查指导原则(2013年修订版)81 防褥疮气床垫产品注册技术审查指导原则82 尿液分析仪产品注册技术审查指导原则83 医用吸引设备产品注册技术审查指导原则84 医用臭氧妇科治疗仪产品注册技术审查指导原则85 血液透析用制水设备产品注册技术审查指导原则86 化学发光免疫分析仪(第二类)产品注册技术审查指导原则87 沉渣分析仪产品注册技术审查指导原则88 视野计产品注册技术审查指导原则89 负压引流装置产品注册技术审查指导原则90 人绒毛膜促性腺激素定性检测试剂(胶体金法)注册申报资料指导原则91 一次性使用无菌手术包类产品注册技术审查指导原则92 一次性使用配药用注射器产品注册技术审查指导原则93 义齿制作用合金产品注册技术审查指导原则94 一次性使用鼻氧管产品注册技术审查指导原则95 便潜血(FOB)定性检测试剂注册申报资料指导原则96 疝修补补片产品注册技术审查指导原则2013年第7/CL0055/2975.html号97 乙型肝炎病毒脱氧核糖核酸定量检测试剂注册技术审查指导原则2013年第3号/CL0057/2976.html98 病原体特异性M型免疫球蛋白定性检测试剂注册技术审查指导原则99 人类免疫缺陷病毒检测试剂临床研究注册技术审查指导原则100 流式细胞仪配套用检测试剂注册技术审查指导原则101 酶联免疫法检测试剂注册技术审查指导原则食药监办械函[2013]3号/CL0027/2434.html102 发光免疫类检测试剂注册技术审查指导原则103 核酸扩增法检测试剂注册技术审查指导原则104 金标类检测试剂注册技术审查指导原则105 生物芯片类检测试剂注册技术审查指导原则106 一次性使用透析器产品注册技术审查指导原则107 凝血分析仪产品注册技术审查指导原则食药监办械函/CL0027/2435.html108 血糖仪产品注册技术审查指导原则[2012]210号109 医用雾化器产品注册技术审查指导原则110 手术电极产品注册技术审查指导原则111 超声多普勒胎儿监护仪产品注册技术审查指导原则112 助听器产品注册技术审查指导原则113 超声洁牙设备产品注册技术审查指导原则114 手术动力设备产品注册技术审查指导原则115 医用分子筛制氧设备产品注册技术审查指导原则116 吻(缝)合器产品注册技术审查指导原则117 麻醉机和呼吸机用呼吸管路产品注册技术审查指导原则118 全瓷义齿用氧化锆瓷块产品注册技术审查指导原则119 流行性感冒病毒核酸检测试剂注册申报资料指导原则食药监办械函/CL0057/1671.html120 流行性感冒病毒抗原检测试剂注册申报资料指导原则[2011]540号121 注射泵产品注册技术审查指导原则食药监办械函[2011]187号/CL0027/2314.html122 红外乳腺检查仪产品注册技术审查指导原则/CL0027/2313.html 123 磁疗产品注册技术审查指导原则/CL0027/2282.html 124 电子血压计(示波法)产品注册技术审查指导原则/CL0027/2312.html 125 3A类半导体激光治疗机产品注册技术审查指导原则/CL0027/2311.html 126 牙科综合治疗机产品注册技术审查指导原则/CL0027/2319.html 127 电动病床产品注册技术审查指导原则/CL0027/2310.html 128 一次性使用手术衣产品注册技术审查指导原则/CL0027/2317.html 129 定制式义齿产品技术审查指导原则/CL0027/2316.html 130 天然胶乳橡胶避孕套产品注册技术审查指导原则/CL0027/2285.html 131 一次性使用真空采血管产品注册技术审查指导原则/CL0027/2315.html132 超声理疗设备产品注册技术审查指导原则/CL0027/2284.html133 角膜塑形用硬性透气接触镜说明书编写指导原则食药监办械函[2011]143号/CL0055/1419.html134 肿瘤标志物类定量检测试剂注册申报资料指导原则食药监办械函[2011]116号/CL0057/1395.html135 体外诊断试剂分析性能评估(准确度—回收试验)技术审查指导原则/CL0057/1394.html136 体外诊断试剂分析性能评估(准确度—方法学比对)技术审查指导原则/CL0057/1393.html137 牙科种植体(系统)产品注册技术审查指导原则/CL0055/1392.html 138 一次性使用输注器具产品注册技术审查指导原则/CL0055/1391.html 139 无源植入性医疗器械货架寿命申报资料指导原则/CL0055/1390.html 140 同种异体植入性医疗器械病毒灭活工艺验证技术审查指导原则/CL0055/1388.html 141 接触镜护理产品注册技术审查指导原则/CL0055/1387.html142 乳房植入体产品注册技术审查指导原则/CL0055/1386.html 143 关于硅橡胶充填式人工乳房产品注册有关问题的通知/CL0055/1355.html144 组织工程医疗产品研究及申报相关要求国食药监械[2007]762号/CL0055/1353.html145 自测用血糖监测系统注册申报资料指导原则食药监办械函[2010]438号/CL0057/1246.html146 植入式心脏起搏器食药监办械函[2010]279号/CL0056/1162.html147 医用X射线诊断设备(第三类)148 影像型超声诊断设备(第三类)149 无源植入性和动物源性医疗器械注册申报资料指导原则食药监办械函[2009]519号/CL0055/1198.html150 X射线诊断设备(第二类)产品注册技术审查指导原则食药监办械函/CL0027/752.html151 骨科外固定支架产品注册技术审查指导原则[2009]95号152 气管插管产品注册技术审查指导原则153 一次性使用无菌导尿管产品注册技术审查指导原则154 胃管产品注册技术审查指导原则155 心电图机产品注册技术审查指导原则156 高强超声聚焦治疗机有关技术要求国食药监械[2003]222号/CL0056/96.html。

序号名称文件号地址1 白蛋白测定试剂(盒)注册技术审查指导原则2016年第29号/WS01/CL0087/145956.html2 糖化血红蛋白测定试剂盒(酶法)注册技术审查指导原则3 乳酸脱氢酶测定试剂盒注册技术审查指导原则4 促甲状腺素检测试剂注册技术审查指导原则5 甘油三酯测定试剂盒注册技术审查指导原则6 唾液酸检测试剂盒(酶法)注册技术审查指导原则7 β2-微球蛋白检测试剂盒(胶乳增强免疫比浊法)注册技术审查指导原则8 碱性磷酸酶测定试剂盒注册技术审查指导原则(2016年修订版)2016年第28号/WS01/CL0087/145955.html9 人绒毛膜促性腺激素检测试剂(胶体金免疫层析法)注册技术审查指导原则(2016修订版)10 C反应蛋白测定试剂盒注册技术审查指导原则(2016年修订版)11 大便隐血(FOB)检测试剂盒(胶体金免疫层析法)注册技术审查指导原则(2016年修订版)12 缺血修饰白蛋白测定试剂盒注册技术审查指导原则(2016年修订版)13 肌酸激酶测定试剂(盒)注册技术审查指导原则(2016年修订版)14 医学图像存储传输软件(PACS)注册技术审查指导原则2016年第27号/WS01/CL0087/145954.html15 正压通气治疗机注册技术审查指导原则16 大型蒸汽灭菌器注册技术审查指导原则17 腹膜透析机注册技术审查指导原则18 医用内窥镜冷光源注册技术审查指导原则19 振动叩击排痰机注册技术审查指导原则20 磁疗产品注册技术审查指导原则(2016年修订版)2016年第22号/WS01/CL0087/144721.html21 电子血压计(示波法)注册技术审查指导原则(2016年修订版)22 X射线诊断设备(第二类)注册技术审查指导原则(2016年修订版)23 尿液分析仪注册技术审查指导原则(2016年修订版)24 半自动化学发光免疫分析仪注册技术审查指导原则(2016年修订版)25 凝血分析仪注册技术审查指导原则(2016年修订版)26 生化分析仪注册技术审查指导原则(2016年修订版)27 血糖仪注册技术审查指导原则(2016年修订版)28 血液透析用制水设备注册技术审查指导原则(2016年修订版)29 牙科综合治疗机注册技术审查指导原则(2016年修订版)30 医用雾化器注册技术审查指导原则(2016年修订版)31 助听器注册技术审查指导原则(2016年修订版)32 自动尿液有形成分分析仪注册技术审查指导原则(2016年修33 高频手术设备注册技术审查指导原则2016年第21号/WS01/CL0087/144620.html34 医用X射线诊断设备(第三类)注册技术审查指导原则(2016年修订版)35 植入式心脏起搏器注册技术审查指导原则(2016年修订版)36 脉搏血氧仪设备临床评价技术指导原则37 治疗呼吸机注册技术审查指导原则38 强脉冲光治疗仪注册技术审查指导原则39 可吸收止血产品注册技术审查指导原则2016年第7号/WS01/CL0087/143162.html40 腹腔、盆腔外科手术用可吸收防粘连产品注册技术审查指导原则41 透明质酸钠类面部注射填充材料注册技术审查指导原则42 一次性使用膜式氧合器注册技术审查指导原则2016年第6号/WS01/CL0087/143160.html43 α-氰基丙烯酸酯类医用粘合剂注册技术审查指导原则44 丙型肝炎病毒核糖核酸测定试剂技术审查指导原则2015年第93号/WS01/CL1107/136485.html45 过敏原特异性IgE抗体检测试剂技术审查指导原则46 人乳头瘤病毒(HPV)核酸检测及基因分型试剂技术审查指导原则47 全自动化学发光免疫分析仪技术审查指导原则48 医疗器械软件注册技术审查指导原则2015年第50号/CL0056/3810.html49 乙型肝炎病毒基因分型检测试剂技术审查指导原则2015年第32号/CL0057/3808.html50 影像型超声诊断设备新技术注册技术审查指导原则2015年第33号/CL0056/3809.html51 雌激素受体、孕激素受体抗体试剂及检测试剂盒技术审查指导原则2015第11号/CL0057/3553.html52 植入式心脏电极导线产品注册技术审查指导原则 2014第10号 /CL0056/2965.html53 医用控温毯产品注册技术审查指导原则 2014年第7号 /CL0027/2969.html54 电动洗胃机产品注册技术审查指导原则 55 医用电子体温计产品注册技术审查指导原则 56 脉搏血氧仪产品注册技术审查指导原则 57 牙科手机产品注册技术审查指导原则58 C 反应蛋白定量检测试剂盒产品注册技术审查指导原则 59 缺血修饰白蛋白测定试剂产品注册技术审查指导原则 60 肌酸激酶测定试剂盒产品注册技术审查指导原则 61 碱性磷酸酶检测试剂盒产品注册技术审查指导原则 62 医用口罩产品注册技术审查指导原则 63 一次性使用引流管产品注册技术审查指导原则64 护脐带产品注册技术审查指导原则65 一次性医用喉罩产品注册技术审查指导原则66 一次性使用皮肤缝合器产品注册技术审查指导原则67 牙科树脂类充填材料产品注册技术审查指导原则2014年第6号/CL0055/2971.html68 一次性使用避光输液器产品注册技术审查指导原则69 一次性使用血液分离器具产品注册技术审查指导原则70 血液透析浓缩物产品注册技术审查指导原则71 金属接骨板内固定系统产品注册技术审查指导原则72 心脏射频消融导管产品注册技术审查指导原则2014年第5号/CL0056/2970.html73 软性亲水接触镜说明书编写指导原则2014年第3号/CL0055/2972.html74 硬性角膜接触镜说明书编写指导原则75 医用磁共振成像系统注册技术审查指导原则2014年第2号/CL0056/2973.html76 弓形虫、风疹病毒、巨细胞病毒、单纯疱疹病毒抗体及G型免疫球蛋白抗体亲合力检测试剂技术审查指导原则77 肿瘤个体化治疗相关基因突变检测试剂技术审查指导原则78 药物滥用检测试剂技术审查指导原则79 红外线治疗设备产品注册技术审查指导原则2013年第8号/CL0027/2974.html80 中频电疗产品注册技术审查指导原则(2013年修订版)81 防褥疮气床垫产品注册技术审查指导原则82 尿液分析仪产品注册技术审查指导原则83 医用吸引设备产品注册技术审查指导原则84 医用臭氧妇科治疗仪产品注册技术审查指导原则85 血液透析用制水设备产品注册技术审查指导原则86 化学发光免疫分析仪(第二类)产品注册技术审查指导原则87 沉渣分析仪产品注册技术审查指导原则88 视野计产品注册技术审查指导原则89 负压引流装置产品注册技术审查指导原则90 人绒毛膜促性腺激素定性检测试剂(胶体金法)注册申报资料指导原则91 一次性使用无菌手术包类产品注册技术审查指导原则92 一次性使用配药用注射器产品注册技术审查指导原则93 义齿制作用合金产品注册技术审查指导原则94 一次性使用鼻氧管产品注册技术审查指导原则95 便潜血(FOB)定性检测试剂注册申报资料指导原则96 疝修补补片产品注册技术审查指导原则2013年第7/CL0055/2975.html号97 乙型肝炎病毒脱氧核糖核酸定量检测试剂注册技术审查指导原则2013年第3号/CL0057/2976.html98 病原体特异性M型免疫球蛋白定性检测试剂注册技术审查指导原则99 人类免疫缺陷病毒检测试剂临床研究注册技术审查指导原则100 流式细胞仪配套用检测试剂注册技术审查指导原则101 酶联免疫法检测试剂注册技术审查指导原则食药监办械函[2013]3号/CL0027/2434.html102 发光免疫类检测试剂注册技术审查指导原则103 核酸扩增法检测试剂注册技术审查指导原则104 金标类检测试剂注册技术审查指导原则105 生物芯片类检测试剂注册技术审查指导原则106 一次性使用透析器产品注册技术审查指导原则107 凝血分析仪产品注册技术审查指导原则食药监办械函/CL0027/2435.html108 血糖仪产品注册技术审查指导原则[2012]210号109 医用雾化器产品注册技术审查指导原则110 手术电极产品注册技术审查指导原则111 超声多普勒胎儿监护仪产品注册技术审查指导原则112 助听器产品注册技术审查指导原则113 超声洁牙设备产品注册技术审查指导原则114 手术动力设备产品注册技术审查指导原则115 医用分子筛制氧设备产品注册技术审查指导原则116 吻(缝)合器产品注册技术审查指导原则117 麻醉机和呼吸机用呼吸管路产品注册技术审查指导原则118 全瓷义齿用氧化锆瓷块产品注册技术审查指导原则119 流行性感冒病毒核酸检测试剂注册申报资料指导原则食药监办械函/CL0057/1671.html120 流行性感冒病毒抗原检测试剂注册申报资料指导原则[2011]540号121 注射泵产品注册技术审查指导原则食药监办械函[2011]187号/CL0027/2314.html122 红外乳腺检查仪产品注册技术审查指导原则/CL0027/2313.html 123 磁疗产品注册技术审查指导原则/CL0027/2282.html 124 电子血压计(示波法)产品注册技术审查指导原则/CL0027/2312.html 125 3A类半导体激光治疗机产品注册技术审查指导原则/CL0027/2311.html 126 牙科综合治疗机产品注册技术审查指导原则/CL0027/2319.html 127 电动病床产品注册技术审查指导原则/CL0027/2310.html 128 一次性使用手术衣产品注册技术审查指导原则/CL0027/2317.html 129 定制式义齿产品技术审查指导原则/CL0027/2316.html 130 天然胶乳橡胶避孕套产品注册技术审查指导原则/CL0027/2285.html 131 一次性使用真空采血管产品注册技术审查指导原则/CL0027/2315.html132 超声理疗设备产品注册技术审查指导原则/CL0027/2284.html133 角膜塑形用硬性透气接触镜说明书编写指导原则食药监办械函[2011]143号/CL0055/1419.html134 肿瘤标志物类定量检测试剂注册申报资料指导原则食药监办械函[2011]116号/CL0057/1395.html135 体外诊断试剂分析性能评估(准确度—回收试验)技术审查指导原则/CL0057/1394.html136 体外诊断试剂分析性能评估(准确度—方法学比对)技术审查指导原则/CL0057/1393.html137 牙科种植体(系统)产品注册技术审查指导原则/CL0055/1392.html 138 一次性使用输注器具产品注册技术审查指导原则/CL0055/1391.html 139 无源植入性医疗器械货架寿命申报资料指导原则/CL0055/1390.html 140 同种异体植入性医疗器械病毒灭活工艺验证技术审查指导原则/CL0055/1388.html 141 接触镜护理产品注册技术审查指导原则/CL0055/1387.html142 乳房植入体产品注册技术审查指导原则/CL0055/1386.html 143 关于硅橡胶充填式人工乳房产品注册有关问题的通知/CL0055/1355.html144 组织工程医疗产品研究及申报相关要求国食药监械[2007]762号/CL0055/1353.html145 自测用血糖监测系统注册申报资料指导原则食药监办械函[2010]438号/CL0057/1246.html146 植入式心脏起搏器食药监办械函[2010]279号/CL0056/1162.html147 医用X射线诊断设备(第三类)148 影像型超声诊断设备(第三类)149 无源植入性和动物源性医疗器械注册申报资料指导原则食药监办械函[2009]519号/CL0055/1198.html150 X射线诊断设备(第二类)产品注册技术审查指导原则食药监办械函/CL0027/752.html151 骨科外固定支架产品注册技术审查指导原则[2009]95号152 气管插管产品注册技术审查指导原则153 一次性使用无菌导尿管产品注册技术审查指导原则154 胃管产品注册技术审查指导原则155 心电图机产品注册技术审查指导原则156 高强超声聚焦治疗机有关技术要求国食药监械[2003]222号/CL0056/96.html。
无菌医疗器械货架有效期验证中容器和密封系统完整性试验替代无菌试验注册技术审查指导原则(征求意见稿)一、前言无菌医疗器械是指不含有活的微生物的医疗器械,包括最终灭菌医疗器械和无菌加工医疗器械。
医疗器械货架有效期是指保证医疗器械终产品正常发挥预期功能的期限,一旦超过医疗器械的货架有效期,就意味着该器械可能不再满足已知的性能指标,发挥预期功能,在使用中具有潜在的风险。
本指导原则系对无菌医疗器械货架有效期验证中容器和密封系统完整性试验替代无菌试验的注册申报资料的一般性要求,未涉及其他技术要求。
对于产品其他技术要求有关注册申报资料的准备,注册申请人还需参考相关的法规和指导性文件。
如有其他法规和指导性文件涉及某类医疗器械货架有效期的具体规定,建议注册申请人结合本指导原则一并使用。
本指导原则系对注册申请人和审查人员的产品有效期研究资料中容器和密封系统完整性试验替代无菌试验注册技术审查方面的指导性文件,不包括注册审批所涉及的行政事项,亦不作为法规强制执行。
如果有能够满足相关法规要求的其他方法,也可采用,但应提供详细的研究资料和验证资料。
注册申请人应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
二、适用范围本指导原则适用范围仅限于无菌医疗器械货架有效期验证中容器和密封系统完整性试验替代无菌试验的相关注册申报资料的准备。
本指导原则不包括以下情况:1)注册申报资料中产品检测报告的无菌试验;2)用于灭菌过程的定义、确认和维护的无菌试验;3)生产企业日常生产过程中的无菌试验。
三、基本要求对于以无菌状态供应的医疗器械,注册申请人应指定一个经过验证的确定的货架有效期。
医疗器械货架有效期的验证试验类型通常可分为实时稳定性试验和加速稳定性试验两类。
医疗器械的实时稳定性试验和加速稳定性试验应同时进行。
无源可重复使用医疗器械使用寿命验证方案从无菌与非无菌两个角度来讨论无源医疗器械的注册检验。
①首先,两者都需要进行生物学检测,ISO10993系列标准是医疗器械生物学评价通用标准。
根据接触时长、接触类型等选择需要进行的生物学检测项目,一般需要2-3个月时间。
②其次,两者都需要适当的标准进行性能检测。
无源医疗器械的主要类别有:眼科产品、口腔产品、外科植入类产品、手术器械和穿刺器械及生育器具。
比如手术衣,在进行CE认证过程中需要采用EN13795-1标准进行性能检测,性能检测周期为15天左右。
③有效期验证/货架寿命试验。
医疗器械货架寿命关乎着医疗器械产品正常发挥,并起到保护医疗器械的作用。
货架的寿命到头,就预示着该器械不再具有性能指标与其功能,在使用中可能会发生不可预知的风险。
加速稳定性试验和实时稳定性试验这两种试验是医疗器械货架寿命的主要验证方式。
后者的试验周期比较长,在注册的时候,可采用加速稳定性试验进行临时货架寿命的确认。
④灭菌验证,这项验证方式主要针对无菌医疗器械,根据产品性质需要选择适当的灭菌工艺。
主要的灭菌方式有三类,分别是:湿热灭菌、辐照灭菌、EO灭菌。
送检的前提下就是灭菌验证。
灭菌工艺确认好之后,就要对产品进行杀菌,后续再进行生物学、包装、性能验证。
以EO灭菌验证为例,其周期一般在80天左右。
⑤无菌医疗器械,需要对产品包装性能进行验证。
不同类型的包装需要根据实际选择包装验证试验,常见的包装测试标准有:密封强度试验:ASTMF88/F88M-15、密封完整性:ASTMF1886/F1886M-16、染色渗漏试验:ASTMF1929-15、无菌屏障系统试验:DIN58953-6:2016等。
⑥无菌医疗器械,还需要模拟运输验证。
保证产品在运输过程中导致破损,导致产品不合格。
附件4:无源植入性医疗器械货架寿命申报资料指导原则一、前言医疗器械货架寿命具有保持医疗器械终产品正常发挥预期功能的重要作用,一旦超过医疗器械的货架寿命,就意味着该器械可能不再具有已知的性能指标及预期功能,在使用中具有潜在的风险。
为进一步明确对无源植入性医疗器械产品注册申报资料的技术要求,指导申请人/生产企业对无源植入性医疗器械货架寿命有关注册申报资料进行准备,特制定本指导原则。
无源非植入性医疗器械有关货架寿命注册申报资料的准备可根据实际情况参照执行。
本指导原则系对无源植入性医疗器械货架寿命的一般性要求,未涉及其他技术要求。
对于产品其他技术要求有关注册申报资料的准备,申请人/生产企业还需参考相关的法规和指导性文件。
如有其他法规和指导性文件涉及某类医疗器械货架寿命的具体规定,建议申请人/生产企业结合本指导原则一并使用。
本指导原则系对申请人/生产企业和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行。
如果有能够满足相关法规要求的其他方法,也可采用,但应提供详细的研究资料和验证资料。
申请者/生产企业应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
二、适用范围本指导原则主要适用于无源植入性医疗器械货架寿命的研究及相关注册申报资料的准备。
三、基本要求(一)货架寿命影响因素影响医疗器械货架寿命的因素主要包括外部因素和内部因素。
此处列举了部分与无源植入性医疗器械密切相关的影响因素,但不仅限于以下内容:外部因素主要包括:1.储存条件,例如温度、湿度、光照、通风情况、气压、污染等;2.运输条件,例如运输过程中的震动、冲撞;3.生产方式,采用不同方式生产的同一医疗器械产品可能具有不同的货架寿命;4.生产环境,如无菌医疗器械生产场所的洁净度、温度和湿度、微生物及悬浮粒子负荷等;5.包装,例如在不同尺寸容器中包装的产品可能具有不同的货架寿命;6.原辅材料来源改变的影响,如采购单位、采购批号改变;7.其他影响因素,如生产设备改变的影响及设备所用清洗剂、模具成型后不清洗的脱模剂的影响。
内部因素一般包括:1.医疗器械中各原材料/组件的自身性能,各原材料/组件随时间的推移而发生退化,导致其化学性能、物理性能或预期功能的改变,进而影响医疗器械整体性能。
如某些高分子材料和组合产品中的药物、生物活性因子等。
2.医疗器械中各原材料/组件之间可能发生的相互作用。
3.医疗器械中各原材料/组件与包装材料(包括保存介质,如角膜接触镜的保存液等)之间可能发生的相互作用。
4.生产工艺对医疗器械中各原材料/组件、包装材料造成的影响,如生产过程中采用的灭菌工艺等。
5.医疗器械中含有的放射性物质和其放射衰变后的副产物对医疗器械中原材料/组件、包装材料的影响。
6.无菌包装产品中微生物屏障的保持能力。
内部因素和外部因素均可不同程度地影响医疗器械产品的技术性能指标,当超出允差后便可造成器械失效。
由于影响因素很多,生产企业不可能将全部影响医疗器械货架寿命的因素进行规避,但应尽可能将各因素进行有效控制,使其对医疗器械技术性能指标造成的影响降至最低。
需要强调的是,并不是所有的医疗器械均需要有一个确定的货架寿命。
当某一医疗器械的原材料性能和包装材料性能随时间推移而不会发生显著性改变时,则可能没有必要确定一个严格的货架寿命,而当某一医疗器械的稳定性较差或临床使用风险过高时,其货架寿命则需要进行严格的验证。
对于以无菌状态供应的无源植入性医疗器械,生产企业应指定一个经过验证的确定的货架寿命。
(二)货架寿命验证过程医疗器械货架寿命的验证贯穿该器械研发的整个过程,生产企业应在医疗器械研发的最初阶段考虑其货架寿命,并在产品的验证和改进过程中不断进行确认。
首先,生产企业要为医疗器械设定保证运输、储存和预期功效的货架寿命。
其次,生产企业需对用于生产和包装医疗器械的材料、组件和相关生产工艺,以及涉及到的参考资料进行全面评估。
如必要,还需进行实验室验证和调整生产工艺。
生产企业根据评估结果设计医疗器械的货架寿命验证方案,并依据方案所获得的验证结果确定该医疗器械的货架寿命。
如验证结果不能被生产企业所接受,则需对其进行改进,并于改进后重新进行验证。
最后,生产企业需要制定严格的质量体系文件以确保产品在货架寿命内进行储存、运输和销售。
生产企业应认真保存医疗器械货架寿命验证过程中涉及的各种文件和试验数据,以便在申请注册时和对货架寿命进行重新评价时提供详细的支持性资料。
(三)货架寿命验证内容1.验证试验类型医疗器械货架寿命的验证试验类型通常可分为加速稳定性试验和实时稳定性试验两类。
(1)加速稳定性试验加速稳定性试验是指将某一产品放臵在外部应力状态下,通过考察应力状态下的材料退化情况,利用已知的加速因子与退化速率关系,推断产品在正常储存条件下的材料退化情况的试验。
加速稳定性试验设计是建立在假设材料变质所涉及的化学反应遵循阿列纽斯(Arrhenius)反应速率函数基础上的。
该函数以碰撞理论为基础,确认化学反应产生变化的反应速率的增加或降低按照以下公式进行:()e kt A dt dq r /φ-==r :反应进行的速率;A :材料的常数(频率因子);φ:表观活化能(eV );k :波尔兹曼常数(0.8617×10-4eV/K );t :绝对温度。
大量化学反应的研究结果表明温度升高或降低10℃会导致化学反应速率增加一倍或减半。
则可根据阿列纽斯反应速率函数建立加速老化简化公式:Q ))((1010T T RT AAT RT AA -=AAT :加速老化时间;RT :实时老化时间;Q 10:温度升高或降低10℃的老化系数;T AA :加速老化温度;T RT :正常储存条件下温度。
上述公式反映了加速稳定性试验中加速老化时间与对应的货架寿命的关系。
其中,Q 10一般设定为2。
当生产企业对医疗器械和包装的材料的评估资料不齐备时,Q 10可保守设定为1.8。
如生产企业在加速稳定性试验中设定的Q 10大于2,则应同时提供详细的相关研究资料。
此外,设定较高的加速老化温度可减少加速稳定性试验的时间。
但是,由于较高的温度可能导致医疗器械原材料/组件和包装材料的性质发生改变或引发多级或多种化学反应,造成试验结果的偏差。
因此,加速老化温度一般不应超过60℃。
如生产企业在加速稳定性试验中设定了更高的加速老化温度,亦应提供详细的相关研究资料。
需要说明的是,当医疗器械的原材料/组件在高温状态下易发生退化和损坏时,则不应采用加速稳定性试验验证其货架寿命。
(2)实时稳定性试验实时稳定性试验是指将某一产品在预定的储存条件下放臵,直至监测到其性能指标不能符合规定要求为止。
实时稳定性试验中,生产企业应根据产品的实际生产、运输和储存情况确定适当的温度、湿度、光照等条件,在设定的时间间隔内对产品进行检测。
由于中国大部分地区为亚热带气候,推荐验证试验中设定的温度、湿度条件为:25℃±2℃,60%RH±10%RH。
无源植入性医疗器械的实时稳定性试验和加速稳定性试验应同时进行。
实时稳定性试验结果是验证产品货架寿命的直接证据。
当加速稳定性试验结果与其不一致时,应以实时稳定性试验结果为准。
2.验证试验检测/评价项目无论加速稳定性试验还是实时稳定性试验,生产企业均需在试验方案中设定检测项目、检测方法及判定标准。
检测项目包括产品自身性能检测和包装系统性能检测两方面。
前者需选择与医疗器械货架寿命密切相关的物理、化学检测项目,涉及产品生物相容性可能发生改变的医疗器械,需进行生物学评价。
如适用,可采用包装封口完整性检测用于替代无菌检测。
后者则包括包装完整性、包装强度和微生物屏障性能等检测项目。
其中,包装完整性检测项目包括染色液穿透法测定透气包装的密封泄漏试验、目力检测和气泡法测定软性包装泄漏试验等;包装强度测试项目包括软性屏障材料密封强度试验、无约束包装抗内压破坏试验和模拟运输试验等。
生产企业在试验过程中应设立多个检测时间点(一般不少于3个)对无源植入性医疗器械进行检测。
可采用零点时间性能数据作为检测项目的参照指标。
3.进行验证试验的产品医疗器械货架寿命验证试验应采用与常规生产相同的终产品进行。
验证的医疗器械应至少包括三个代表性批次的产品,推荐采用连续三批。
生产企业可对试验产品进行设计最差条件下的验证试验以保证试验产品可代表最恶劣的生产情况,如进行一个标准的灭菌周期后,附加一个或多个灭菌周期,或采用几种不同的灭菌方法。
4.验证试验中采用的统计处理方法生产企业应在验证试验方案中设定每一检测项目的检测样品数量以确保检测结果具有统计学意义,并在试验报告中提供相关信息。
(四)参考标准建议医疗器械生产企业尽可能采用国家标准、行业标准和公认的国际标准中规定的方法/措施对其医疗器械产品货架寿命进行验证,以减少验证结果的偏差,提高验证结论的准确性。
附件中列举了可能在货架寿命验证过程中涉及的部分标准,但不仅限于所列内容。
(五)注册时应提交的技术文件生产企业在无源植入性医疗器械首次注册时需提供详细的货架寿命验证资料,一般包括以下内容:1.与申请注册产品货架寿命相关的基本信息。
包括该医疗器械原材料/组件、包装材料、生产工艺、灭菌方法(如涉及)、货架寿命、储存运输条件等;2.生产企业在该医疗器械货架寿命验证过程中对相关影响因素的评估报告;3.实时稳定性试验的试验方案及试验报告,同时提供试验方案中检测项目、检测判定标准、检测时间点及检测样本量的确定依据和相关研究资料;4.如适用,可提供加速稳定性试验的试验方案和试验报告,同时提供加速稳定性试验的试验方案中检测项目、检测判定标准、加速老化参数、检测时间点及检测样本量的确定依据和相关研究资料;5.包装工艺验证报告及包装、密封设备的详述;6.生产企业认为应在注册时提交的其他相关支持性资料。
生产企业可在申请注册产品的货架寿命技术文件中使用其生产的其他医疗器械产品的货架寿命研究资料及验证资料,但应同时提供二者在原材料、包装材料、生产工艺、灭菌方法(如涉及)等与货架寿命相关的信息对比资料和二者在货架寿命方面具有等同性的论证资料。
四、名词解释1. 植入性医疗器械(Implantable Medical Device)是指任何通过外科手术达到下列目的的医疗器械:全部或部分插入人体或自然腔道中,或为替代上表皮或眼表面;此类医疗器械,通过外科手段在术后臵留体内30天以上,并只能通过内科或外科手段取出。
(该定义不适用于有源植入性医疗器械)2.货架寿命(Shelf Life)是指医疗器械形成终产品后能够发挥拟定作用的时间段。