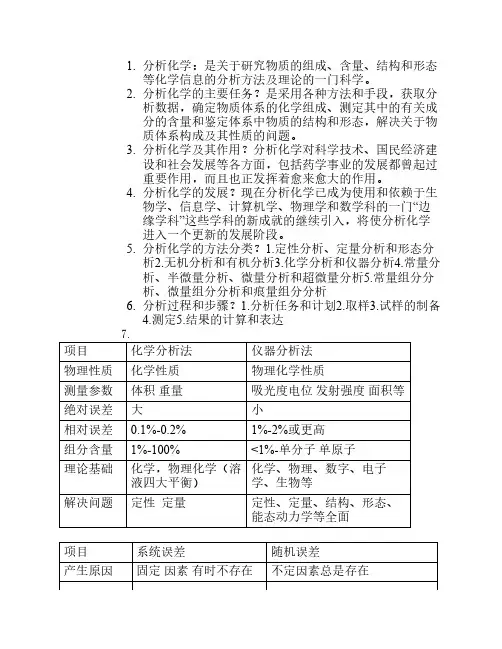
分析化学
- 格式:docx
- 大小:25.67 KB
- 文档页数:7
E=xi-uEr=(xi Nhomakorabeau)/u
同样的绝对误差,当被测定的量较大时,相对误差就比较小,测定的准确度也就比较高。 准确的是指测定平均值与真值接近的程度。 2、 偏差:指个别测定值 xi 与几次测定结果的平均值 x 之间的差别。可用绝对偏差 di 和相 对偏差 dr 表示。
di =xi-x dr =
xi − x x
多组分分析 双波长分光光度法 有机化合物电子跃迁类型
原子吸收光谱法(Atomic Absorption Spectrometry AAS) 根据原子外层电子跃迁所产生的光谱进行分析的方法,称为原子光谱法。 包括原子发射光谱法,原子吸收光谱法,原子荧光光谱法。 原子吸收光谱法, 是基于测量试样所产生的原子蒸气中基态原子对其特征谱线的吸收, 从而 定量测定化学元素的方法。 公式:A = KC A:吸光度 K:一个常数 C:浓度 原子吸收光谱仪: 光源:空心阴极管。作用是发射待测元素的特征光谱,以供试样吸收之用。 火焰原子化系统: 将试样中待测元素转变成基态原子蒸汽。 待测元素由化合物离解成基态原 子的过程,称为原子化过程。 火焰原子化装置:包括雾化器和燃烧器两部分。 雾化器:将试液雾化。目前普遍采用同心雾化器。 燃烧器:形成火焰,使进入火焰的试样微粒原子化。 火焰的作用是提供一定的能量,促使试样雾滴蒸发,干燥并经过热离解或还原作用,产生大 量基态原子。火焰超过温度,激发态原子将增多,电离度增大,基态原子减少,这对原子吸 收不利。 助燃气:空气、氧气或者氧化亚氮。 燃气:乙炔、丙烷、氢气等。 燃气和助燃气的流量决定火焰的状态。 非火焰原子化装置:石墨炉原子化器。 分光系统:主要由色散元件,凹面镜,狭缝组成。 检测系统:由检测器(光电倍增管) ,放大器,读数,记录系统等组成。 定量分析方法: 1、 标准曲线法 2、 标准加入法 灵敏度:用矫正曲线的斜率 S 表示,或者用特征浓度来表示。 检出极限:指仪器能以适当的置信度检出的待测元素的最小浓度或最小量。
分析方法一般分为化学分析和仪器分析。 分析化学:以化学反应为基础的分析方法。例如重量分析法和滴定分析法。 仪器分析: 借助光电仪器测量试样溶液的光学性质, 电学性质等物理或者物理化学性质来求 出待测组分含量的方法。例如吸光光度法,红外/紫外吸收光谱法,原子吸收光谱法,色谱 法等。 光学分析法:吸光光度法,红外吸收光谱分析法,紫外吸收光谱分析法,发射光谱分析法, 原子吸收光谱分析法,荧光分析法。 电化学分析法:电重量分析法,极谱分析法。 物理及物理化学分析方法:色谱法 误差及分析数据的统计处理: 1、 误差:测定值 xi 与真值 u 之差,误差的大小可用绝对误差 E 和相对误差 Er 表示。
滴定分析(Titrimetric Analysis) 使用滴定管将一种已知准确浓度的试剂溶剂即标准溶液滴加到待测物的溶液中, 直到待测组 分恰好完全反应,然后根据标准溶液的浓度和消耗的体积,算出待测组分的含量。 滴定:titration 化学计量点:stoichiommetric point 指示剂:indicator 滴定终点:end point 终点误差:end point error 滴定分析法分为:酸碱滴定法 沉淀滴定法 络合滴定法 氧化还原滴定法 标准溶液的配制: 直接法 间接法 (*标定) 滴定度:指与每毫升标准溶液相当的被测组分的质量,用 T 表示。 酸碱滴定法(Acid-Base Titration) PH 的计算 标准溶液的配制:Hcl NaOH 络合滴定法(Complexometry) 络合剂 EDTA:乙二胺四乙酸(H4 Y) 直接法 返滴法 置换滴法 间接法 氧化还原滴定法(Oxidation Reduction Titrimetry) 氧化还原指示剂:二苯胺磺酸钠,亚甲基蓝等。 高锰酸钾法:KMnO4 配制: 可 用 于 双 氧 水 , 钙 , 铁 , 有 机 物 , COD ( 水 样 中 化 学 耗 氧 量 ) 的 测 定 (* 含 Cl−高的工业废水要用重络酸钾法)。 重铬酸钾法:K 2 Cr2 O7 配制:易于提纯,可以准确称取一定质量的重络酸钾制成一定浓度的标准溶液。 可用于CODCr 碘量法:I2 硫代硫酸钠标准溶液;碘标准溶液
× 100%
算术平均偏差 相对平均偏差 标准偏差:表示数据集的离散程度。值越大,数据集越分散,值越小,数据集越集中。 精密度:在确定条件下,将测试方法实施几次,求出所得结果之间的一致程度。可常用重复 性和再现性表示。 实验结果首先要求精密度高, 才有保证有准确的结果, 但高的精密度也不一定有高的准确性。 3、 误差的分类 系统误差(可测误差) 随机误差(偶然误差) 系统误差产生的原因: 方法不完善造成的方法误差; 实验试剂不纯,测量仪器缺陷等造成的误差。 操作人员操作不当或操作偏见造成的人为误差。 空白试验,回收试验。 随机误差产生的原因: 由一些无法控制的不确定因素所引起的,如环境温度,湿度,电压,污染情况等。 随机误差服从正态分布。 4、 分析结果数据处理 可疑数据的取舍:Grubbs 法,Q 值检验法 平均值与标准值的比较:检验方法的准确性,即用已知含量的标准试样进行试验。 5、 有效数字的保留
溴酸钾法: 其他
重量分析法和沉淀分析法(Gravimetry and Precipitation Titrametry) 1、 测定Cl− 用K 2 CrO4 做指示剂,用硝酸银标准溶液进行滴定。
吸光光度法(Spectrophotometry) 基于物质对光的选择性吸收而建立的分析方法称为吸光光度法, 包括比色法, 可见分光光度 法,紫外分光光度法。 将不同波长的光透过某一固定浓度和厚度的有色溶液, 测量每一波长下有色溶液对光的吸收 程度(即吸光度 A) ,然后以波长为横坐标,以吸光度 A 为纵坐标作图,即可得到吸收曲线 (吸收光谱) 。它描述物质对不同波长光的吸收能力。吸收曲线中有一高峰,相应的波长称 为最大吸收波长,用λmax 表示。 公式:A = − log10 T = log10 A:吸光度 T:透光度 T =
2
3、 峰基宽度(peak width at peak base,Wb ) 塔板理论(plate theory) n = 速率理论:H = A + u + Cu 分离度 R(resolution) R= 2(t R 2 − t R 1 ) Wb (2) + Wb (1)
B
L H
载气种类及流速的选择: 载气流速一般用柱前载气的体积流速来表示,用转子流量计来测量。 柱温的选择: 柱长和柱内径的选择: 进样量和进样时间的选择: 汽化温度的选择: 毛细管柱气相色谱法(Capillary Column Chromatography) 气相色谱检测器 作用是将色谱柱分离后的各组分,按其物理,化学特性转换为易测量的电信号 E 氢火焰离子化检测器(FID) 几种常用定量方法: 1、 归一化法 2、 内标法 3、 外标法
I0 I I I0
= abc
I0 :入射光强度 I:透射光强度
a:吸收系数 b:以 cm 为单位 如果 c 以g ∙ L−1 为单位,则 a 的单位为L ∙ g−1 ∙ cm−1 ;如果 c 以mol ∙ L−1 为单位,则 a 的单位 为L ∙ mol−1 ∙ cm−1 ,称为摩尔吸收系数,用符号 k 表示。则 A=kbc k 是吸光物质在特定波长和溶剂的情况下的一个特征常数,数值上等于浓度为 1 摩尔每升吸 光物质在 1cm 光程中的吸光度,是物质吸光能力的量度。k 值愈大,方法的灵敏度愈高。 k=Ma M:物质的摩尔质量 偏离比尔定律的原因:1、仪器不能真正提供单色光。 2、 高浓度溶液粒子间作用力的影响。 风光光度计 光源:可见光常用钨丝灯 320~2500nm 为光源;在近紫外区测定时常用氢等,氚等产生 180~375nm 的连续光谱。 单色器: 将光源发出的连续光谱分解为单色光的装置, 由棱镜或者光栅等色散元件及狭缝和 透镜等组成。 吸收池:也称比色皿。可用无色透明,能耐腐蚀性的玻璃比色皿。大多数仪器配有液层厚度 为 0.5cm,1cm,2cm,3cm 等的一套长方形比色皿。使用时应注意比色皿透光面垂直于光束方 向,指纹,油渍或者其他沉积物都会影响其投射特性,因此应注意保持比色皿的光洁。 检测系统:测量吸光度时,是将光强度转换为电流来进行测量的,这种光电转换器称为光电 检测器。 显色反应 将待测组分转变成有色化合物的反应叫做显色反应, 与待测组分形成有色化合物的试剂称为 显色剂。 显色反应可分为络合反应和氧化还原反应。 显色剂过量愈多, 愈有利于待测组分形成有色配合物, 但是显色剂过量加入也会引起副作用, 对测定不利。 有时候光度分析中, 共存离子如本身有颜色或者与显色剂作用生成有色化合物, 将干扰测定, 应该先消除共存离子的干扰。 参比溶液的选择:用纯溶剂或者空白溶剂(蒸馏水)做参比溶液。
气相色谱分析法(Gas Chromatography,GC) 色谱法中起分离作用的柱称为色谱柱, 固定在柱内的填充物称为固定相, 沿着柱流动的流体 称为流动相。用液体作为流动相称为液相色谱,用气体作为流动相称为气相色谱。 气相色谱分析流程: 气相色谱的流动相称为载气,常用H2 ,N2 等。载气由高压钢瓶供给,经过调压,干燥净化 后, 用气流调节阀调节并控制载气流速至所需值, 到达净化室。 试样用注射器由进样口注入, 在气化室瞬间汽化,被载气带入色谱柱进行分离,分离后随载气先后进入检测器,记录器记 录下信号。 气相色谱柱主要有两种,一种是内装固定相的填充柱,另一种是内壁涂固定液的毛细管柱。 后者因阻力小,可以做得很长,因而柱的分离能力强,分析速度快。 在一定温度下,组分在两相间分配达到平衡时的浓度比,称为分配系数。 固定相: 气固色谱一般用表面具有一定火星的吸附剂作为固定相, 常用的有非极性的活性炭, 极性的 Al2 O3 ,氢键型的硅胶。 固定相的选择: 1、 分离非极性组分,一般选用非极性固定液,各组分按沸点次序流出色谱柱,沸点低的先 出峰。 2、 分离极性组分, 选用极性固定液, 各组分按极性大小顺序流出色谱柱, 极性小的先出峰。 3、 分离极性和非极性的混合物,一般选用极性固定液,非极性组分先出峰。 气相色谱流出曲线及有关术语 在气相色谱分析中将以组分浓度为纵坐标, 留出时间为横坐标, 绘得组分浓度随时间变化的 曲线称为色谱图。 基线(base line) :只有载气通过检测器时响应信号的记录即为基线。 保留值(retention value) :表示试样中各组分在色谱柱内停留时间的数值,通常用时间或对 应的载气体积来表示。 1、 用时间表示的保留值 保留时间(retention time,t R ),指待测组分从进样到柱后出现浓度最大值时所需的时间。 死时间(dead time,t M ),指不与固定相作用的气体的保留时间。 调整保留时间(adjusted retention time, t R′ ),指扣除了死时间的保留时间。 2、 用体积表示的保留值 保留体积(retention volume, VR ),指从进样到柱后出现待测组分浓度最大值时所通过的载气 体积。VR = t R ∙ F0 F0 为色谱柱出口处载气流速。 死体积(dead volume, VM ) 调整保留体积(adjusted retention volume, VR ′) 区域宽度(peak width) 1、 标准偏差(standard deviation,σ) 2、 半峰宽(peak width at half height,Y1 )
同样的绝对误差,当被测定的量较大时,相对误差就比较小,测定的准确度也就比较高。 准确的是指测定平均值与真值接近的程度。 2、 偏差:指个别测定值 xi 与几次测定结果的平均值 x 之间的差别。可用绝对偏差 di 和相 对偏差 dr 表示。
di =xi-x dr =
xi − x x
多组分分析 双波长分光光度法 有机化合物电子跃迁类型
原子吸收光谱法(Atomic Absorption Spectrometry AAS) 根据原子外层电子跃迁所产生的光谱进行分析的方法,称为原子光谱法。 包括原子发射光谱法,原子吸收光谱法,原子荧光光谱法。 原子吸收光谱法, 是基于测量试样所产生的原子蒸气中基态原子对其特征谱线的吸收, 从而 定量测定化学元素的方法。 公式:A = KC A:吸光度 K:一个常数 C:浓度 原子吸收光谱仪: 光源:空心阴极管。作用是发射待测元素的特征光谱,以供试样吸收之用。 火焰原子化系统: 将试样中待测元素转变成基态原子蒸汽。 待测元素由化合物离解成基态原 子的过程,称为原子化过程。 火焰原子化装置:包括雾化器和燃烧器两部分。 雾化器:将试液雾化。目前普遍采用同心雾化器。 燃烧器:形成火焰,使进入火焰的试样微粒原子化。 火焰的作用是提供一定的能量,促使试样雾滴蒸发,干燥并经过热离解或还原作用,产生大 量基态原子。火焰超过温度,激发态原子将增多,电离度增大,基态原子减少,这对原子吸 收不利。 助燃气:空气、氧气或者氧化亚氮。 燃气:乙炔、丙烷、氢气等。 燃气和助燃气的流量决定火焰的状态。 非火焰原子化装置:石墨炉原子化器。 分光系统:主要由色散元件,凹面镜,狭缝组成。 检测系统:由检测器(光电倍增管) ,放大器,读数,记录系统等组成。 定量分析方法: 1、 标准曲线法 2、 标准加入法 灵敏度:用矫正曲线的斜率 S 表示,或者用特征浓度来表示。 检出极限:指仪器能以适当的置信度检出的待测元素的最小浓度或最小量。
分析方法一般分为化学分析和仪器分析。 分析化学:以化学反应为基础的分析方法。例如重量分析法和滴定分析法。 仪器分析: 借助光电仪器测量试样溶液的光学性质, 电学性质等物理或者物理化学性质来求 出待测组分含量的方法。例如吸光光度法,红外/紫外吸收光谱法,原子吸收光谱法,色谱 法等。 光学分析法:吸光光度法,红外吸收光谱分析法,紫外吸收光谱分析法,发射光谱分析法, 原子吸收光谱分析法,荧光分析法。 电化学分析法:电重量分析法,极谱分析法。 物理及物理化学分析方法:色谱法 误差及分析数据的统计处理: 1、 误差:测定值 xi 与真值 u 之差,误差的大小可用绝对误差 E 和相对误差 Er 表示。
滴定分析(Titrimetric Analysis) 使用滴定管将一种已知准确浓度的试剂溶剂即标准溶液滴加到待测物的溶液中, 直到待测组 分恰好完全反应,然后根据标准溶液的浓度和消耗的体积,算出待测组分的含量。 滴定:titration 化学计量点:stoichiommetric point 指示剂:indicator 滴定终点:end point 终点误差:end point error 滴定分析法分为:酸碱滴定法 沉淀滴定法 络合滴定法 氧化还原滴定法 标准溶液的配制: 直接法 间接法 (*标定) 滴定度:指与每毫升标准溶液相当的被测组分的质量,用 T 表示。 酸碱滴定法(Acid-Base Titration) PH 的计算 标准溶液的配制:Hcl NaOH 络合滴定法(Complexometry) 络合剂 EDTA:乙二胺四乙酸(H4 Y) 直接法 返滴法 置换滴法 间接法 氧化还原滴定法(Oxidation Reduction Titrimetry) 氧化还原指示剂:二苯胺磺酸钠,亚甲基蓝等。 高锰酸钾法:KMnO4 配制: 可 用 于 双 氧 水 , 钙 , 铁 , 有 机 物 , COD ( 水 样 中 化 学 耗 氧 量 ) 的 测 定 (* 含 Cl−高的工业废水要用重络酸钾法)。 重铬酸钾法:K 2 Cr2 O7 配制:易于提纯,可以准确称取一定质量的重络酸钾制成一定浓度的标准溶液。 可用于CODCr 碘量法:I2 硫代硫酸钠标准溶液;碘标准溶液
× 100%
算术平均偏差 相对平均偏差 标准偏差:表示数据集的离散程度。值越大,数据集越分散,值越小,数据集越集中。 精密度:在确定条件下,将测试方法实施几次,求出所得结果之间的一致程度。可常用重复 性和再现性表示。 实验结果首先要求精密度高, 才有保证有准确的结果, 但高的精密度也不一定有高的准确性。 3、 误差的分类 系统误差(可测误差) 随机误差(偶然误差) 系统误差产生的原因: 方法不完善造成的方法误差; 实验试剂不纯,测量仪器缺陷等造成的误差。 操作人员操作不当或操作偏见造成的人为误差。 空白试验,回收试验。 随机误差产生的原因: 由一些无法控制的不确定因素所引起的,如环境温度,湿度,电压,污染情况等。 随机误差服从正态分布。 4、 分析结果数据处理 可疑数据的取舍:Grubbs 法,Q 值检验法 平均值与标准值的比较:检验方法的准确性,即用已知含量的标准试样进行试验。 5、 有效数字的保留
溴酸钾法: 其他
重量分析法和沉淀分析法(Gravimetry and Precipitation Titrametry) 1、 测定Cl− 用K 2 CrO4 做指示剂,用硝酸银标准溶液进行滴定。
吸光光度法(Spectrophotometry) 基于物质对光的选择性吸收而建立的分析方法称为吸光光度法, 包括比色法, 可见分光光度 法,紫外分光光度法。 将不同波长的光透过某一固定浓度和厚度的有色溶液, 测量每一波长下有色溶液对光的吸收 程度(即吸光度 A) ,然后以波长为横坐标,以吸光度 A 为纵坐标作图,即可得到吸收曲线 (吸收光谱) 。它描述物质对不同波长光的吸收能力。吸收曲线中有一高峰,相应的波长称 为最大吸收波长,用λmax 表示。 公式:A = − log10 T = log10 A:吸光度 T:透光度 T =
2
3、 峰基宽度(peak width at peak base,Wb ) 塔板理论(plate theory) n = 速率理论:H = A + u + Cu 分离度 R(resolution) R= 2(t R 2 − t R 1 ) Wb (2) + Wb (1)
B
L H
载气种类及流速的选择: 载气流速一般用柱前载气的体积流速来表示,用转子流量计来测量。 柱温的选择: 柱长和柱内径的选择: 进样量和进样时间的选择: 汽化温度的选择: 毛细管柱气相色谱法(Capillary Column Chromatography) 气相色谱检测器 作用是将色谱柱分离后的各组分,按其物理,化学特性转换为易测量的电信号 E 氢火焰离子化检测器(FID) 几种常用定量方法: 1、 归一化法 2、 内标法 3、 外标法
I0 I I I0
= abc
I0 :入射光强度 I:透射光强度
a:吸收系数 b:以 cm 为单位 如果 c 以g ∙ L−1 为单位,则 a 的单位为L ∙ g−1 ∙ cm−1 ;如果 c 以mol ∙ L−1 为单位,则 a 的单位 为L ∙ mol−1 ∙ cm−1 ,称为摩尔吸收系数,用符号 k 表示。则 A=kbc k 是吸光物质在特定波长和溶剂的情况下的一个特征常数,数值上等于浓度为 1 摩尔每升吸 光物质在 1cm 光程中的吸光度,是物质吸光能力的量度。k 值愈大,方法的灵敏度愈高。 k=Ma M:物质的摩尔质量 偏离比尔定律的原因:1、仪器不能真正提供单色光。 2、 高浓度溶液粒子间作用力的影响。 风光光度计 光源:可见光常用钨丝灯 320~2500nm 为光源;在近紫外区测定时常用氢等,氚等产生 180~375nm 的连续光谱。 单色器: 将光源发出的连续光谱分解为单色光的装置, 由棱镜或者光栅等色散元件及狭缝和 透镜等组成。 吸收池:也称比色皿。可用无色透明,能耐腐蚀性的玻璃比色皿。大多数仪器配有液层厚度 为 0.5cm,1cm,2cm,3cm 等的一套长方形比色皿。使用时应注意比色皿透光面垂直于光束方 向,指纹,油渍或者其他沉积物都会影响其投射特性,因此应注意保持比色皿的光洁。 检测系统:测量吸光度时,是将光强度转换为电流来进行测量的,这种光电转换器称为光电 检测器。 显色反应 将待测组分转变成有色化合物的反应叫做显色反应, 与待测组分形成有色化合物的试剂称为 显色剂。 显色反应可分为络合反应和氧化还原反应。 显色剂过量愈多, 愈有利于待测组分形成有色配合物, 但是显色剂过量加入也会引起副作用, 对测定不利。 有时候光度分析中, 共存离子如本身有颜色或者与显色剂作用生成有色化合物, 将干扰测定, 应该先消除共存离子的干扰。 参比溶液的选择:用纯溶剂或者空白溶剂(蒸馏水)做参比溶液。
气相色谱分析法(Gas Chromatography,GC) 色谱法中起分离作用的柱称为色谱柱, 固定在柱内的填充物称为固定相, 沿着柱流动的流体 称为流动相。用液体作为流动相称为液相色谱,用气体作为流动相称为气相色谱。 气相色谱分析流程: 气相色谱的流动相称为载气,常用H2 ,N2 等。载气由高压钢瓶供给,经过调压,干燥净化 后, 用气流调节阀调节并控制载气流速至所需值, 到达净化室。 试样用注射器由进样口注入, 在气化室瞬间汽化,被载气带入色谱柱进行分离,分离后随载气先后进入检测器,记录器记 录下信号。 气相色谱柱主要有两种,一种是内装固定相的填充柱,另一种是内壁涂固定液的毛细管柱。 后者因阻力小,可以做得很长,因而柱的分离能力强,分析速度快。 在一定温度下,组分在两相间分配达到平衡时的浓度比,称为分配系数。 固定相: 气固色谱一般用表面具有一定火星的吸附剂作为固定相, 常用的有非极性的活性炭, 极性的 Al2 O3 ,氢键型的硅胶。 固定相的选择: 1、 分离非极性组分,一般选用非极性固定液,各组分按沸点次序流出色谱柱,沸点低的先 出峰。 2、 分离极性组分, 选用极性固定液, 各组分按极性大小顺序流出色谱柱, 极性小的先出峰。 3、 分离极性和非极性的混合物,一般选用极性固定液,非极性组分先出峰。 气相色谱流出曲线及有关术语 在气相色谱分析中将以组分浓度为纵坐标, 留出时间为横坐标, 绘得组分浓度随时间变化的 曲线称为色谱图。 基线(base line) :只有载气通过检测器时响应信号的记录即为基线。 保留值(retention value) :表示试样中各组分在色谱柱内停留时间的数值,通常用时间或对 应的载气体积来表示。 1、 用时间表示的保留值 保留时间(retention time,t R ),指待测组分从进样到柱后出现浓度最大值时所需的时间。 死时间(dead time,t M ),指不与固定相作用的气体的保留时间。 调整保留时间(adjusted retention time, t R′ ),指扣除了死时间的保留时间。 2、 用体积表示的保留值 保留体积(retention volume, VR ),指从进样到柱后出现待测组分浓度最大值时所通过的载气 体积。VR = t R ∙ F0 F0 为色谱柱出口处载气流速。 死体积(dead volume, VM ) 调整保留体积(adjusted retention volume, VR ′) 区域宽度(peak width) 1、 标准偏差(standard deviation,σ) 2、 半峰宽(peak width at half height,Y1 )