
MEGA的使用
产生背景及简介
随着不同物种基因组测序的快速发展,产生了大量的DNA序列信息,这时就需要一种简便而快速的统计分析工具来对这些数据进行有效的分析,以提取其中包含的大量信息。MEGA就是基于这种需求开发的。MEGA 软件的目的就是提供一个以进化的角度从DNA和蛋白序列中提取有用的信息的工具,并且,此软件可以免费下载使用。
现在我们使用的是MEGA4的版本。它主要集中于进化分析获得的综合的序列信息。使用它我们可以编辑序列数据、序列比对、构建系统发育树、推测物种间的进化距离等。此软件的输出结果资源管理器允许用户浏览、编辑、打印输入所得到的结果而且所得到的结果具有不同形式的可视化效果。此外,该软件还能够得出不同序列间的距离矩阵,这是他不同与其他分析软件的地方。在计算矩阵方面有一些自己的特点:
1.推测序列或者物种间的进化距离
2.根据MCL(Maximum Composite Likeliood method)的方法构建系统发育树
3.考虑到了不同碱基替换的不同的比率,考虑到了碱基转换和颠换的差别。
4.随时可以使用标注:所以的结果输入都可以使用标注,而且标注的内容
可以被保存,复制。
具体使用
我们以分析20个物种的血红蛋白为例来具体说明此软件的具体使用情况。一.启动程序
1.运行环境:在Windows 95/98, NT, ME, 2000, XP, vista等操作系统下均可使用。
2.下载安装:可以直接登陆https://www.doczj.com/doc/dd7064586.html,进行下载安装,另外还可以
从https://www.doczj.com/doc/dd7064586.html,/tools/phylogeny.php中的链接进去。
3.双击桌面快捷方式图标,进入主界面;或者从开始菜单,单击图标启
动。
二.序列分析。
1.启动
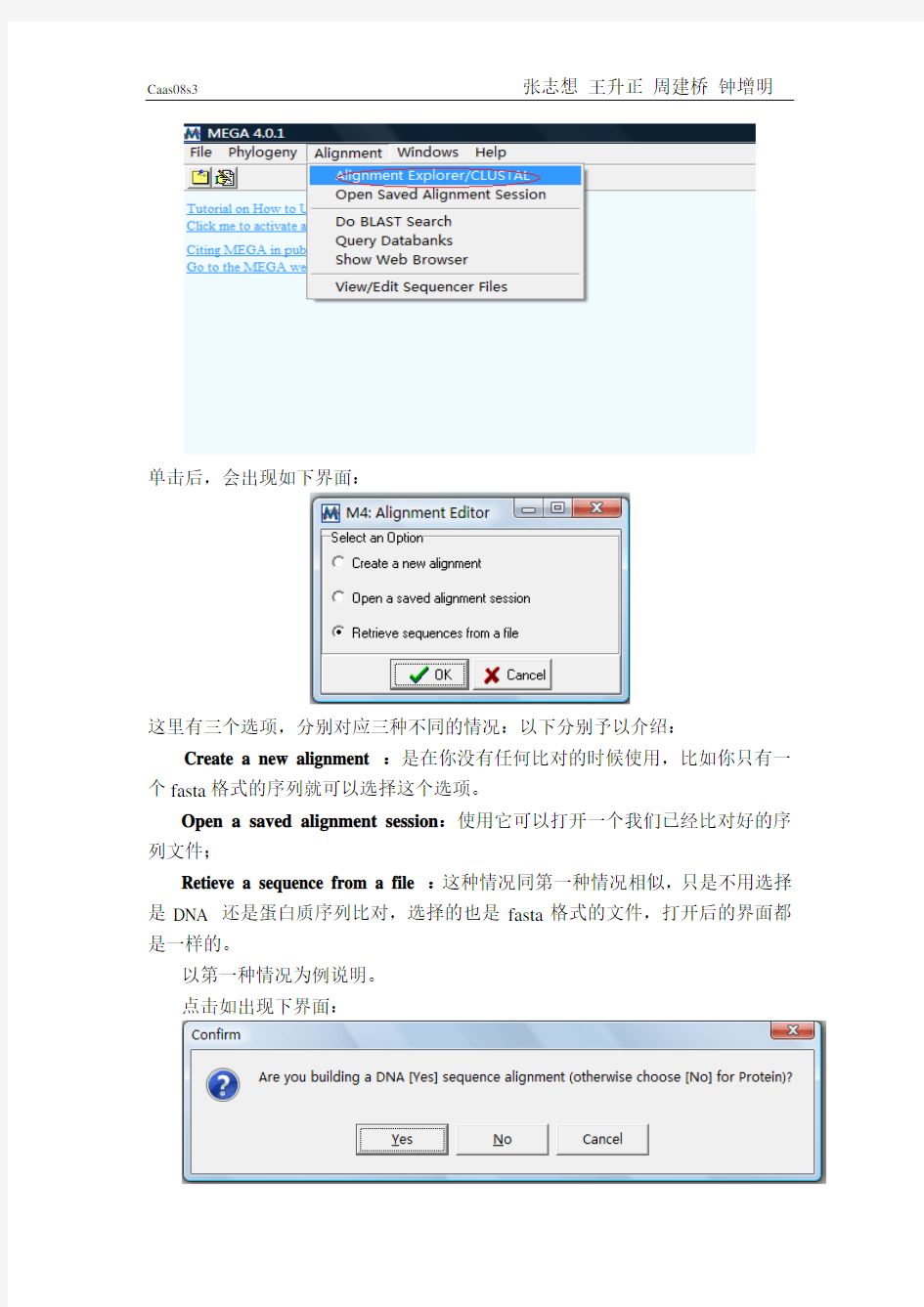
单击后,会出现如下界面:
这里有三个选项,分别对应三种不同的情况:以下分别予以介绍:Create a new alignment :是在你没有任何比对的时候使用,比如你只有一个fasta格式的序列就可以选择这个选项。
Open a saved alignment session:使用它可以打开一个我们已经比对好的序列文件;
Retieve a sequence from a file :这种情况同第一种情况相似,只是不用选择是DNA 还是蛋白质序列比对,选择的也是fasta格式的文件,打开后的界面都是一样的。
以第一种情况为例说明。
点击如出现下界面:
这里我们分析的是蛋白序列所以选择No。点击出现如下界面:
然后从data菜单选择输入数据文件如图:
选择你保存的fasta格式序列后就会出现:
下面介绍菜单的使用:
Data:
Creat a new :创建一个新的数据比对文件,也就是说当我们比对完一组后,想接着比对另一组,那么使用它就可以不用退出直接把数据文件导入;
Open:打开先前已经比对并保存好的文件,它包含两个子菜单:retive sequence from file和saved aligment session ;
Close: 关闭当前的比对数据文件;
Save session:保存当前比对结果,可以给比对的结果一个文件名;
Export alignment:将当前的序列比对结果输出到指定文件,有两种输入格式可供选择:MGTA和FASTA.
DNA sequence:使用它来选择输入的数据DNA序列,这里需要说明的是如果你输入的数据是氨基酸序列的话,比对窗口只显示一个标签,若是DNA序列
的话则显示两个标签,一个是DNA序列的,另一个是氨基酸序列的。如图:
Protein sequences:选择输入的氨基酸序列,选择后,所以的位点就被当作氨基酸残基位点来对待。
Translate/untranslate:只有比对的序列是编码蛋白的DNA序列的时候才可用。它可以根据指定的遗传密码表将DNA序列翻译成特定的氨基酸序列。
Select genetic code table:使用它将编码蛋白的DNA翻译成特定的蛋白序列。
Reverse complement:将选择的一整行的DNA序列变为与之互补配对碱基序列。
Exit alignment explorer:退出序列比对的资源管理窗口。
Edit菜单:
使用这个菜单可以对我们的比对序列进行想要的一些编辑工作具体为
Undo:撤销上一步操作;
Copy:复制;cut:剪切;Paste:粘贴;前面三个操作都可以只针对一个碱基或氨基酸残基也可以是一段甚至是整个序列;
Delete:从比对表格中删除一段序列;
Delete gaps:去掉序列中的空缺;
Insert blank sequence:重新插入一空行;标签和序列都是空的;
Insert sequence from file:从已保存的文件中插入新的序列;
Select sites:选择一列序列,与点击比对表上方的灰白空格作用类似;
Select sequence:选择一行序列,与点击比对表格左侧的标签名作用类似;
Select all:全选;
Allow base editing:只读保护,只有选择后才能对序列进行编辑操作,否则所以的序列为只读格式,不能进行任何编辑操作。
Search菜单:
用来快捷查找序列中的标记未定或者目的碱基或残基。
Find motif:选择后出现如下对话框:
输入你想要查看的一小段序列。找到后会以黄色标出;
Find next:在序列的下游查找目的序列片段;
Find preious:在序列的上有查找目的序列片段;
Find marked sites:查找标记位点;
Highlight motif:突出标记已经选择的位点。
Web菜单
这个菜单提供一个链接Genbank的入口,可以在网上直接做Blast搜索。当手上没有准备好要比对的序列时,可以直接去网上搜索。
Query gene banks:开启NCBI的主页;
Do blast search:开启NCBI BLAST主页;
Show browser:开启网页浏览器。
Sequencer菜单
此菜单下只有一个子菜单:edit sequencer file,用来打开一个打开文件对话框,此对话框可以打开一个sequencer data file,一旦打开,这个文件就在trace data file viewer/editor的对话框中展示出来。这个编辑窗口允许你查看和编辑automatd DNA sequencer 产生的trace data。它可以阅读和编辑ABI和Staden格式文件并且序列可以直接被导入到序列比对窗口或被上传到网页浏览器做blast搜索。
Display菜单:
这个菜单相对简单,主要用来调整工具栏。
Toolbars:工具栏菜单,它包含一些子菜单,选择后就会出现在比对的窗口中;
Use colors:将不同的位点以不同的颜色显示;
Background color:选择后位点的显示与位点一样的背景颜色;
Font:字体对话框,通过选择来调整窗口中的序列字符的大小。
最后,结合实例来介绍alignment 菜单
Mark/unmark site:在比对的表格中标记或者不标记一个单一位点,一次每条序列只能被标记一个位点,不同序列间的位点你可以选择同一列的,也可以是错开的,要根据自己的目的进行选择。选择标记后的序列可以使用align marked sites 进行比对分析。
Align marked sites:比对标记的序列,在这里如果在两个或多个序列间标记了不在一列的位点重新比对后会出现空格。如图:
Unmarked all sites:把所以标记的位点去标记;
Delete gap-only site:去掉序同是空格的一列;这在多序列比对前很有用。
Auto-fill gaps:使用空格补齐不同长度的序列。
Align by clustalw:此软件整合了clustalw程序,这也是它的方便之处,选择要比对的序列后点击会出现下面的对话框:
一般参数:
DNA/protein weight matrix:选择不同的加权矩阵;
Residue-specific penalties:特殊氨基酸罚分。在序列比对的过程中特异氨基酸可能增加或减少罚分值,比如:富含甘氨酸的区段比富含缬氨酸的区段更可能有空格出现,因而他们的罚分不同。
Hydrophilic penalties:如果有连续的5个或者更多的亲水性氨基酸的话,他们倾向于出现空格,这些区段很可能出现环状或卷曲,因此罚分不一样。
Gap separation distance:参数设置来尽可能降低空格之间离的太近的机会,小于指定数值的空格罚分要多余其他的,这不能避免出现相邻空格,只能降低他们出现的频率。
Use negative matrix:使用负性矩阵,
Delay divergent cutoff:若一条序列相似性低于设定的百分值将推迟比对。
当一切参数都设定好了之后就点击OK就可以进行比对了,中间出现一个过
度对话框,等一下就出现如下界面:
注意:以上介绍的许多操作菜单在窗口的上放有快捷操作按钮。如上图标记内。
比对结束后,可以将结果保存(data/save session/),以供构建系统发育树使用。另外,如果不保存直接关闭,系统跳出一个对话框:
下面这个是序列数据管理的管理界面,此外我们还可以通过主界面上的data/open data路径打开,效果是一样的,注意这里打开的只能是刚才保存的后缀是.MEG的文件。
几个功能菜单:
下面就着重介绍一下序列数据窗口的一些具体使用:
这个窗口用来展示比对后的序列数据,这里提供了许多的功能菜单用来查看
序列比对后的数据统计结果或者来选择想要的子序列。
Data 菜单
Write data to file:导入序列打开窗口;
Translate/untranslate:将蛋白编码序列翻译成蛋白序列,或者再转变成核酸序列;
Selected genetic code table:打开select genetic code对话框,从这个对话框可以选择编辑或者添加遗传密码表;
Setup/select genes and domains:打开sequence data organizer对话框,在这个对话框里可以定义和编辑基因和结构域。
使用这个窗口可以查看,定义,和选择结构域和基因,并且标记单个的位点。具体使用这里不作详细介绍。
Setup/select taxa and groups:打开一个可以编辑分类和定义分类组的对话框:
这个窗口分为两个子窗口,左边的是分类组,显示不同的分组情况,右边的是未分组窗口显示还没有归入任何一个组群的分类。中间和下边是一些操作键,通过他们我们可以建立新的组,如果你将所以的分类都归入到不同的组里,并且给予组名,你们在序列数据窗口中就会在物种名字后边显示他所属的组名。
Quite data viewer:退出界面。
Display 菜单:
Show only selected sequence:只显示你所选择的感兴趣的序列;
Use identical symbol:将一列中绝大部分相同等碱基或氨基酸字符用点来代替;
Color cells:将序列中连续的一致的碱基或者氨基酸给以相同的颜色背景以区别显示;
Sort sequences:将显示的分类以不同的方式排序,可以根据序列名字、组名来排序;
Restore input order:将经过修改的序列顺序回复到刚打开时的样子;
Show sequence name:显示序列的名字,不选则隐藏;
Grouped:显示组名;
Change font:更改显示的字体格式。
Highlight 菜单
这里的子菜单大部分都显示在工具栏里,如图所示:
Statistics 菜单
Nucleotide composition:当序列为核酸时可用。计算每条序列中的不同的碱基百分比;
Nucleotide pair frenquencies:只有当序列为核酸时可用。
Codon usage:只有序列为编码蛋白的核酸序列时可用。计算出codon usage 的百分比和RSCU(relative synonymous codon usage)值;
Amino acid composition:当序列为氨基酸序列或编码蛋白的核酸序列时可用。计算每条序列氨基酸残基的百分比;并且跳出一个显示窗口,在这个窗口中可以进行许多操作:
可以得到的这一数据保存到文件中;还可以打印出来;还以直接分析统计所
得到的结果,查看每一行等。具体大家可以自己摸索;
Use all selected sites:保证上面的分析统计是在选择所有的序列下进行的,不考虑被标记的位点。
三.从以上大家应该可以粗略的了解到这个软件的强大而又方便的序列比对分析的功能。下面再简要介绍主页面上的几个菜单的使用。
Distances菜单:
相关原理:两条序列间的进化距离是通过计算两条序列间碱基或氨基酸替换得来的,推测进化距离是研究分子进化、构建系统发育树和推测物种分化时间的基础,这个软件中包括了绝大部分广泛使用的推测进化距离的方法。值得提出的是,该软件还使用解析公式和bootstrap的方法来评价出现的错误。
该软件所包括的方法大致可被分为三类:核酸;同义—非同义替换;氨基酸。
1)核酸:序列是核酸和核酸之间的比较,计算编码蛋白和非编码蛋白的核
酸序列间的进化距离,主要有两种方法: No. of differences 和
p-distance还包括许多的模型:Jukes-Cantor Model 、Tajima-Nei
Model、Kimura 2-Parameter Model、Tamura 3-Parameter Model、
Tamura-Nei Model、Maximum Composite Likelihood Model等,可以根
据需要进行不同的选择。
2)同义-非同义替换:序列是编码子和编码子之间的比较,所以只能用来计
算编码蛋白的序列。常用的模型有: Nei-Gojobori Method 、 Modified Nei-Gojobori Method 、Li-Wu-Luo Method 、 Pamilo-Bianchi-Li
Method、Kumar Method 等。
3)氨基酸类:序列间是氨基酸残基之间的比较。能够用来计算氨基酸序列
间以及编码蛋白的核酸间的距离,编码蛋白的核酸在比对的时候自动被
翻译成氨基酸序列进行比较。常用的模型有:Poisson Model、Equal Input Model、 Dayhoff and JTT Models。
Choose model…:选择模型,选择跳出一个距离模型的选项窗口:
在这个窗口里,model选项是选择推测进化距离的随机模型的,可以通过单击绿色小方框进行选择。Pattern among lineages:只有当距离模型选定后才可用;rates among sites:允许位点间存在不同的替换率。选好后单击OK 即可。
Compute pairwise:单击出现上面类似的对话框:
Compute:选择是只计算进化距离还是选择计算同时进行评价。选择后者会出现standard error computation by选项,通过这一选项选择解析公式或者bootstrap method来评价结果的好坏。Gaps and missing data:在计算开始前选择去除所有包含比对空格和失意的位点;另外,最初你也可以保留这些位点,在必要的时候在去掉。Labled sites:只有当一些或者全部位点有相关标签时才可用。点击绿色方框,就可以看到包括选择标签的位点,如果你选择这些位点的话,这些位点就最先从数据中提出来。选好后compute出现以下窗口:
这是一个比对后的距离矩阵窗口,这个窗口包括很多不同的功能菜单,来调节显示的内容。File菜单中有一个子菜单是Show Analysis Description:显示计算所用的不同的选项,这些信息可以被保存或者打印出来。
Average Menu:这里面有个子菜单Overall单击会显示比对的总体平均距离。
Distance菜单中其他的子菜单操作同上类似只是内容略有不同,具体可自行摸索。
Phylogeny
Phylogeny选项中有以下子菜单:
其中Construct Phylogeny和Bootstrap Test of Phylogeny基本一致,其中后者给出了在计算过程中的出现的概率。
下面用以下21条血红蛋白序列来对各种方法做一个简单的介绍。
AC ID 中文名
P08850 ACCGE 苍鹰
P07417 AEGMO 黑秃鹰
P18969 AILFU 小熊猫
P18970 AILME 大熊猫
P84216 BATEA 鳐鱼
P02020 LEPPA 南美肺鱼
P01999 ALLMI 美洲鳄
P18972 CALAR 银绢绒猴
P69905 HUMAN 人
P63108 MACMU 猕猴
P21768 MACSI 斯里兰卡猴
P18975 PANLE 狮
P63109 PANON 美洲虎
P63110 PANPO 远东豹
P69906 PANPA 倭黑猩猩
P09420 SPECI 长尾黄鼠
P11750 SPEPA 北极黄鼠
P07428 XENTR 非洲爪蟾
P02010 VIPAS 蝰蛇
P14389 PTEAL 黑妖狐蝠
P14390 PTEPO 灰头狐蝠
最大简约法Maximum Parsimony,使用的运算法则是branch-and bound的检索方法。得到的是无根树。这种方法在序列非常相似以及序列数目较小的情形下较适用(构建21条序列的进化树时,在几种方法中花费的时间最长)。
在实际运行得到拓扑图之后,上面有两个选项,点击Original tree,可以选择查看计算所得到的所有结构树。
点击Bootstrap consensus tree得到我们所需要的结果
点击,则可以指定树根。
点击和,可以调整树枝的位置。
点击按钮,得到的是给出了相对遗传距离的进化树
点击按钮,得到结构树标出了整个树枝长度代表的遗传距离。
邻接法Neighbor Joining。当所考虑的谱系间进化速率可变时,邻接法特别适用。邻接法能给出枝长最小平方估计的序列,即能最真实的反映序列间的真实距离。邻接法得到的进化树也是无根树。邻接法有6种计算方法,分别是No. of Differences、p-distance、Poisson Correction、Equal Input、PAM Matrix (Dayhoff)、JTT Matrix (Jones-Taylor-Thornton)。通常选
择p-distance。得到的系统结构树为:
HUMAN
PANPA CALAR MACMU MACSI SPECI SPEPA PTEAL PTEPO AI LFU AI LME PANLE
PANON
PANPO
ALLMI ACCGE AEGMO
VI PAS
XENTR
BATEA
LEPPA
100
100
1
009182
89
5376100977153
89
75
73100
71
1000.02
最小进化法Minimum Evolution 。该方法和邻接法基本相似,在此不作介绍。 算术平均的非加权对群法UPGMA 。它假设沿着进化树分支的变化速率为一个常数,而距离近似为非加权的。UPGMA 法由计算关系最近序列间的枝长开始,然后计算序列对与下一个序列对间的距离平均值,不断重复直到所有序列都被包括在树中。如果树枝间的突变率不一致时,UPGMA 法将导致一个错误的树,因此该法现在已基本不用。
Relative Rate Tests
点击Tajima’s Test ,得到下面的对话框。
MEGA软件的使用 引言 现代分子生物学所积累的数据库(如美国国家生物信息中心建立的GeneBank等)隐含着大量的生物系统学和生物进化的有用信息。计算机软件是挖掘这些知识宝藏的最有效的工具,而且这些数据库不断快速扩展,信息量十分庞大。因此,如果没有计算机软件的帮助,我们简直无法开战分子系统学和分子进化方面的研究工作。同样,这些数据分析方法和软件在古DNA研究中是必不可少的。 因为有着坚实的分子进化和人类遗传学基础,序列比对分析已经成为重构物种和基因家族进化历史,估算分子进化速率、推断基因和基因组进化过程中自然选择力量的强度等的必不可少的方法和手段。计算机的应用和统计学的介入大大简化这些工作。在这些背景下,Sudhir Kumar、Koichiro Tamura和Masatonshi Nei 和在上世纪九十年代初就发展了Mega遗传分析软件,并不断改进。现在公布了3.0版,增添很多新功能,并使软件使用者能在线取得帮助。 Mega(Molecular Evolutionary Genetics Analysis)是一个界面友好、操作简便、功能强大的分子进化遗传分析软件,也是文献中经常用到的分析软件。尤其是,Mega的新版本对使用界面做了优化,并有改进了许多统计学和遗传学算法,其支持的文件格式很多,而且可以直接从测序图谱中读取序列。另外,Mega 软件还内嵌了一个Web浏览器,能直接登录NCBI网站。 Mega软件操作起来很方便,其界面与传统的Windows程序界面很像,即使初学者也很易上手。 Mega软件功能十分强大,尤其在计算遗传距离、构建分子系统树方面。Mega 软件提供多种计算距离的模型,包括Jukes-Cantor距离模型、Kimura距离模型、Equal-input距离模型、Tamura距离模型、HEY距离模型、Tamura-Nei距离模型、General reversible距离模型、无限制距离模型等。Mega软件可以计算个体之间的遗传距离,还可估算群体间的遗传差异,及群体间的净遗传距离;而其还可以估算一个群体或整个样本的基因分歧度的大小。另外Mega还提供了多种构建分子系统树的方法,包括算术平均的不加权对群法(UPGMA,unweighted pair group method with arithmetic mean),邻接法(NJ,Neighbor-Joining),最大简约法(MP,Maximum Parsimony)、最小进化法(ME,Minimum Evolution)等。在此基础上,Mega软件还提供了对已构建系统树的检验,包括自展法(Bootstrap Method)检验和内部分支检验等。在对于自然选择方面,Mega软件提供了Codon-Based Z 检验、Codon-Based Fisher`s 原样检验t和Tajima中性检验三种方法。总之,Mega 软件提供了构建分子系统树,进行系统发育分析各个方面的计算和分析。 本章将以古DNA数据分析为例,介绍Mega软件的基本原理和方法、使用和操作、以及相关结果的分析。 Mega软件包的下载网址为:https://www.doczj.com/doc/dd7064586.html,
Mega软件输入数据的格式 Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。 首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*.txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来。在与“Title”同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。4)在“#MEGA”和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。这些文字可用来说明诸如作者、分析日期、分析目的等信息。5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。在同一个数据文件里,不能出现数据名相同的序列。在数据名及具体序列中,空格和TAB是被忽略的。6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?”表示,比对时的空缺位点可以用“—”表示。下边是一个数据文件示例: Fig 其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#”,每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。下边是一个数据文件示例: Fig 下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。 Fig 再次,如果数据是测序图谱的形式,直接导入即可。下图是测序图谱示例: Fig MEGA界面及操作 Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。 1、数据的录入及编辑 Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据
MEGA的使用 产生背景及简介 随着不同物种基因组测序的快速发展,产生了大量的DNA序列信息,这时就需要一种简便而快速的统计分析工具来对这些数据进行有效的分析,以提取其中包含的大量信息。MEGA就是基于这种需求开发的。MEGA 软件的目的就是提供一个以进化的角度从DNA和蛋白序列中提取有用的信息的工具,并且,此软件可以免费下载使用。 现在我们使用的是MEGA4的版本。它主要集中于进化分析获得的综合的序列信息。使用它我们可以编辑序列数据、序列比对、构建系统发育树、推测物种间的进化距离等。此软件的输出结果资源管理器允许用户浏览、编辑、打印输入所得到的结果而且所得到的结果具有不同形式的可视化效果。此外,该软件还能够得出不同序列间的距离矩阵,这是他不同与其他分析软件的地方。在计算矩阵方面有一些自己的特点: 1.推测序列或者物种间的进化距离 2.根据MCL(Maximum Composite Likeliood method)的方法构建系统发育树 3.考虑到了不同碱基替换的不同的比率,考虑到了碱基转换和颠换的差别。 4.随时可以使用标注:所以的结果输入都可以使用标注,而且标注的内容 可以被保存,复制。 具体使用 我们以分析20个物种的血红蛋白为例来具体说明此软件的具体使用情况。一.启动程序 1.运行环境:在Windows 95/98, NT, ME, 2000, XP, vista等操作系统下均可使用。 2.下载安装:可以直接登陆https://www.doczj.com/doc/dd7064586.html,进行下载安装,另外还可以 从https://www.doczj.com/doc/dd7064586.html,/tools/phylogeny.php中的链接进去。 3.双击桌面快捷方式图标,进入主界面;或者从开始菜单,单击图标启 动。 二.序列分析。 1.启动
MEGA软件的使用 Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。 1、数据的录入及编辑 Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。 首先,打开Mega程序,有如下图所示的操作界面: 单击工具栏中的“File”按钮,会出现如下图所示的菜单: 从上图可以看出,下拉菜单有“Open Data”(打开数据)、“Reopen Data”(打开曾经打开的数据,一般会保留新近打开的几个数据)、“Close Data”(关闭数据)、“Export Data”(导出数据)、“Conver To MEGA Format”(将数据转化为MEGA格式)、“Text Editor”(数据文本编辑)、“Printer Setup”(启动打印)、“Exit”(退出MEGA程序)。单击“Open Data”选项,会弹出如下菜单:
浏览文件,选择要分析的数据打开,单击“打开”按钮,会弹出如下操作界面: 此程序操作界面,提供了三种选择数据选择:Nucleotide Sequences(核苷酸序列)、Protein Sequences(蛋白质序列)、Pairwise Distance(遗传距离矩阵)。根据输入数据的类型,选择一种,点击“OK”即可。如果选择“Pairwise Distance”,则操作界面有所不同;如下图所示: 根据遗传距离矩阵的类型,如果是下三角矩阵,选择“Lower Left Matrix”即可;如果是上三角矩阵,选择“Upper Right Matrix”即可。点击“OK”按钮,即可导入数据。如果是核苷酸数据,则读完之后,会弹出如下对话框:
MEGA软件构建系统发育树 摘要:以白色念珠菌属下面的十个种的18s RNA 为例,构建系统发育树来说明MEGA 软件的使用方法。 1背景简介 1.1 MEGA(分子进化遗传分析) MEGA 的全称是Molecular Evolutionary Genetics Analysis。MEGA is an integrated tool for automatic and manual sequence alignment, inferring phylogenetic trees, mining web-based databases, estimating rates of molecular evolution, and testing evolutionary hypotheses. MEGA 可用于序列比对、进化树的推断、估计分子进化速度、验证进化假说等。MEGA 还可以通过网络(NCBI)进行序列的比对和数据的搜索。 最新版本:MEGA 5.1 Beta (软件开发者建议其结果不用于发表文章) 建议下载版本:MEGA 5.05 for Windows and Mac OS。 MEGA 5 has been tested on the following Microsoft Windows? operating systems: Windows 95/98, NT, 2000, XP, Vista, version 7, Linux and Mac OS [1]. MEGA 5.05 可免费下载,只需输入名字及有效,下载会发送至,点击可下载。1.2 系统发育树定义 系统发育树(英文:Phylogenetic tree)又称为演化树(evolutionary tree),是表明被认为具有共同祖先的各物种间演化关系的树。是一种亲缘分支分类方法(cladogram)。在树中,每个节点代表其各分支的最近共同祖先,而节点间的线段长度对应演化距离(如估计的演化时间) 1.3 系统发育树的分类 根据有根和无根来区分:树可分为有根树和无根树两类。有根树是具有方向的
1.准备序列文件 准备fasta格式序列文件(fasta格式:大于号>后紧跟序列名,换行后是序列。举例如下)。每条序列可以单独为一个文件,也可以把所有序列放在同一文件内。 核酸序列: >sequence1_name CCTGGCTCAGGATGAACGCT 氨基酸序列: >sequence2_name MQSPINSFKKALAEGRTQIGF 2.多序列比对 打开MEGA 5,点击Align,选择Edit/Build Alignment,选择Create a new alignment,点击OK。
这时需要选择序列类型,核酸(DNA)或氨基酸(Protein)。 选择之后,在弹出的窗口中直接Ctrl + V粘贴序列(如果所有序列在同一个文件中,即可全选序列,复制)。也可以:点击Edit,选择Insert Sequence From File,选择序列文件(可多选)。
序列文件加载之后,呈蓝色背景(为选中状态)。点击按钮,选择Align DNA (如果是氨基酸序列,则会出现Align Protein)。弹出的窗口中设置比对参数,一般都是采用默认参数即可。点击OK,开始多序列比对。
比对完成后,呈现以下状态。 这时需要截齐两端含有---的序列:选中含有---的序列,按键Delete删除(注意:两端都需要截齐)。截齐之后,保存文件为:filename.mas
3.构建系统进化树 多序列比对窗口,点击Data,选择Phylogenetic Analysis,弹出窗口询问:所用序列是否编码蛋白质,根据实际情况选择Yes或No。此时,多序列比对文件就激活了,可以返回MEGA 5主界面建树了。
如何用MEGA构建进化树 MEGA3、1就是一个关于序列分析以及比较统计得工具包,其中包括有距离建树法与MP建树法;可自动或手动进行序列比对,推断进化树,估算分子进化率,进行进化假设测验,还能联机得Web数据库检索。下载后可直接使用,主要包括几个方面得功能软件:i)DNA与蛋白质序列数据得分析软件。ii)序列数据转变成距离数据后,对距离数据分析得软件。iii)对基因频率与连续得元素分析得软件。iv)把序列得每个碱基/氨基酸独立瞧待(碱基/氨基酸只有0与1得状态)时,对序列进行分析得软件。v)绘制与修改进化树得软件,进行网上blast搜索。 用MEGA构建进化树有以下步骤: 1、16S rDNA测序与参考序列选取 从环境中分离到单克隆,去重复后扩增16S rDNA序列并测序,然后与数据库比对,找到相似度最高得几个序列,确定一下您分离得细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定您分离得到得就就是Blast到得那个,然后找一到两个同科得,再找一到两个同目得,再找一到两个同纲得细菌,把序列全部下下来,以FSATA形式整合在TXT文档中,如 >TS1 GCAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAACACGTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCGGATAGGACCTCGGGA TGCATGTTCCGGGGTGGAAAGGTTTTCCGGTGCAGGATGGGCC >gi|117572706|gb|EF028124、1| Rhodococcus sp、Atl25 16S ribosomal RNAgene,partial sequence CGATTAGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGATTAGTGGCGAACGGGTGAGTAACACGTGGGTGA TCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCGGAT >TS2 TGCAAGTCGAGCGAATGGA TTAAGAGCTTGCTCTTATGAAGTTAGCGGCGGA CGGGTGAGTAACACGTGGGTAACCTGCCCATAAGACTGGGATAACTCCGG GAAACCGGGGCTAATACCGGATAACATTTTGAACTGCATGGTTCGAAATTGAAAGGCGGCTTCGGCTGTCACT >gi|56383044|emb|AJ809498、1|Bacillus cereus partial16S rRNA gene, strainTMW 2、383 GA TGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGAATGGATTAAGAGCTTGCTCTTATGAAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCCATAAGACTGGGA TAACTCCGGGAAACCGGGGCTAA TACCGGATAACA TTTTGAACYGCA TGGTTC…………………………、 …………………………、 参考序列选择有几个原则:a,不选非培养(unclutured)微生物为参比;b,所选参考序列要正确,里面无错误碱基;c,在保证同属得前提下,优先选择16S rDNA全长测序或全基因组测序得种;d,每个种属选择一个参考序列,如果自己得序列中同一属得较多,可适当选择两个参考序列。 2、序列比对
来源:https://www.doczj.com/doc/dd7064586.html,/ Arduino Mega2560 简介 Arduino Mega2560也是采用USB接口的核心电路板,它最大的特点就是具有多达54路数字输入输出,特别适合需要大量IO接口的设计。Mega2560的处理器核心是ATmega2560,同时具有54路数字输入/输出口(其中16路可作为PWM输出),16路模拟输入,4路UART接口,一个16MHz晶体振荡器,一个USB口,一个电源插座,一个ICSP header 和一个复位按钮。Arduino Mega2560也能兼容为Arduino UNO设计的扩展板。Arduino Mega2560已经发布到第三版,与前两版相比有以下新的特点: 在AREF处增加了两个管脚SDA和SCL,支持I2C接口;增加IOREF 和一个预留管脚,将来扩展板将能兼容5V和3.3V核心板。 改进了复位电路设计。 USB接口芯片由ATmega16U2替代了ATmega8U2。
概要 ?处理器 ATmega2560 ?工作电压 5V ?输入电压(推荐) 7-12V ?输入电压(范围) 6-20V ?数字IO脚 54 (其中16路作为PWM输出)?模拟输入脚 16 ?IO脚直流电流 40 mA ? 3.3V脚直流电流 50 mA
?Flash Memory 256 KB (ATmega328,其中8 KB 用于bootloader) ?SRAM 8 KB ?EEPROM 4 KB ?工作时钟 16 MHz 电路图和PCB ?电路图https://www.doczj.com/doc/dd7064586.html,/en/uploads/Main/arduino-mega2560 -schematic.pdf ?硬件设计文件(Eagle文件)https://www.doczj.com/doc/dd7064586.html,/en/uploads/Main/arduino-mega25 60-reference-design.zip ?引脚图https://www.doczj.com/doc/dd7064586.html,/en/Hacking/PinMapping2560 电源 Arduino Mega2560可以通过3种方式供电,而且能自动选择供电方式 ?外部直流电源通过电源插座供电。 ?电池连接电源连接器的GND和VIN引脚。 ?USB接口直接供电。 电源引脚说明
用MEGA2做进化树的步骤(图示) 1、打开程序 如下图所示: 2、MEGA2只能打开meg格式的文件,但是它可以把其他格式的多序列比对文件转换过来,我们在这里用aln格式(Clustal的输出文件)转换meg文件。点File:Convert to MEGA Format...打开转换文件对话框 如下图所示:
3、选择文件和转换文件对话框,选择aln文件,点OK 如下图所示: 4、转换好的meg文件,点存盘保存meg文件,meg文件会和aln文件保存在同一个目录 如下图所示: 5、关闭转换窗口,回到主窗口,现在点面板上的“Click me to activate a data file”打开刚才的meg 文件 如下图所示:
6、选择meg文件,点“打开” 如下图所示: 7、程序会自动识别序列的类型,如果识别错误,请手工选择数据类型。然后点OK就行了如下图所示:
8、数据输入之后的样子,窗口下面有序列文件名和类型 如下图所示: 9、现在终于可以开始做Bootstrap验证和进化树了,MEGA的主要功能就是做Bootstrap验证的进化树分析,Bootstrap验证是对进化树进行统计验证的一种方法,可以作为进化树可靠性的一个度量。各种算法虽然不同,但是操作方法基本一致,我们在此以UPGMA方法为例进行演示。点下图所示的菜单项。 如下图所示:
10、...会弹出如下的对话框,在此你可以选择计算参数。 如下图所示: 11、Distance Options标签页中的Models可以下拉,其中有若干个计算距离的方法可以选择,在此默认泊松校验(Poisson Correction)作为计算距离的方法。 如下图所示:
MEGA软件的使用 Mega软件输入数据的格式 Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。 首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*.txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来。在与“Title”同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。4)在“#MEGA”和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。这些文字可用来说明诸如作者、分析日期、分析目的等信息。5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。在同一个数据文件里,不能出现数据名相同的序列。在数据名及具体序列中,空格和TAB是被忽略的。6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?”表示,比对时的空缺位点可以用“—”表示。下边是一个数据文件示例: Fig 其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#”,每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。下边是一个数据文件示例:
Fig 下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。 Fig 再次,如果数据是测序图谱的形式,直接导入即可。下图是测序图谱示例:
MEGA的主界面: 使用步骤 1. 序列导入 可以通过Data导入数据也可以通过file导入数据。 如果打开的文件是比对结果,选择Analyze;如果打开的文件是序列文件,选择Align。 另外双击这些后缀名文件即可自动导入序列,导入后会弹出MEGA比对界面。
如果fasta 序列导入报错,多是因为序列长度不同导致: 如果序列长度不同,可以采用新建文件,将序列文件导入的方法。 步骤:Align → Edit/Build Alignment → create a new alignment → Data → open → Retrieve sequences from File
将复制输入的序列另存输出看看。 步骤:data → Export alignment → fasta format 序列长度都被用横线补齐了。 2. 多序列比对
选择muscle或者clustalw进行比对:clustalw 一般用于DNA ,muscle多用于蛋白。 在比对之前需先选中要进行比对的序列(Shift),还可以对序列或者序列名进行编辑(双击)。比对参数选择:
保存比对文件,进化树分析提供数据。
一般导出的比对结果保存为fasta格式,或者直接点击保存按钮将结果,保存为二进制的mas或meg文件。 3. 构建进化树 导入数据:将刚刚另存的meg 文件重新导入到mega程序中(直接拖入工作界面),并选择构建进化树。 参数选择:参数设置,Bootstrap method一般选择1000~1500;第一次绘图时建议选择500,这样运行速度会比价快,结果合适再调至1000重新进行进化分析。
MEGA构建系统进化树的步骤 1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。如图: 2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图: 。 3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,单击右键,选择delete即可,如图: 。
4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。最后出现一个对话框询问是否打开,选择Yes,如图: 。 5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,
不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图: 在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,
MEGA4的中文使用说明 产生背景及简介 随着不同物种基因组测序的快速发展,产生了大量的DNA 序列信息,这时就需要一种简便而快速的统计分析工具来对这些数据进行有效的分析,以提取其中包含的大量信息。MEGA 就是基于这种需求开发的。MEGA 软件的目的就是提供一个以进化的角度从DNA 和蛋白序列中提取有用的信息的工具,并且,此软件可以免费下载使用。 现在我们使用的是 MEGA4 的版本。它主要集中于进化分析获得的综合的序列信息。使用它我们可以编辑序列数据、序列比对、构建系统发育树、推测物种间的进化距离等。此软件的输出结果资源管理器允许用户浏览、编辑、打印输入所得到的结果而且所得到的结果具有不同形式的可视化效果。此外,该软件还能够得出不同序列间的距离矩阵,这是他不同与其他分析软件的地方。在计算矩阵方面有一些自己的特点: 1. 推测序列或者物种间的进化距离 2. 根据MCL(Maximum Composite Likeliood method)的方法构建系统发育树 3. 考虑到了不同碱基替换的不同的比率,考虑到了碱基转换和颠换的差别。 4. 随时可以使用标注:所以的结果输入都可以使用标注,而且标注的内容可以被保存,复制。 具体使用 我们以分析 20 个物种的血红蛋白为例来具体说明此软件的具体使用情况。 启动程序 1. 运行环境:在Windows 95/98, NT, ME, 2000, XP, vista 等操作系统下均可使用。 2. 下载安装:可以直接登陆https://www.doczj.com/doc/dd7064586.html, 进行下载安装,另外还可以从https://www.doczj.com/doc/dd7064586.html,/tools/phylogeny.php 中的链接进去。 3. 双击桌面快捷方式图标, 进入主界面;或者从开始菜单,单击图标启动。 序列分析 1.启动 单击后,会出现如下界面: