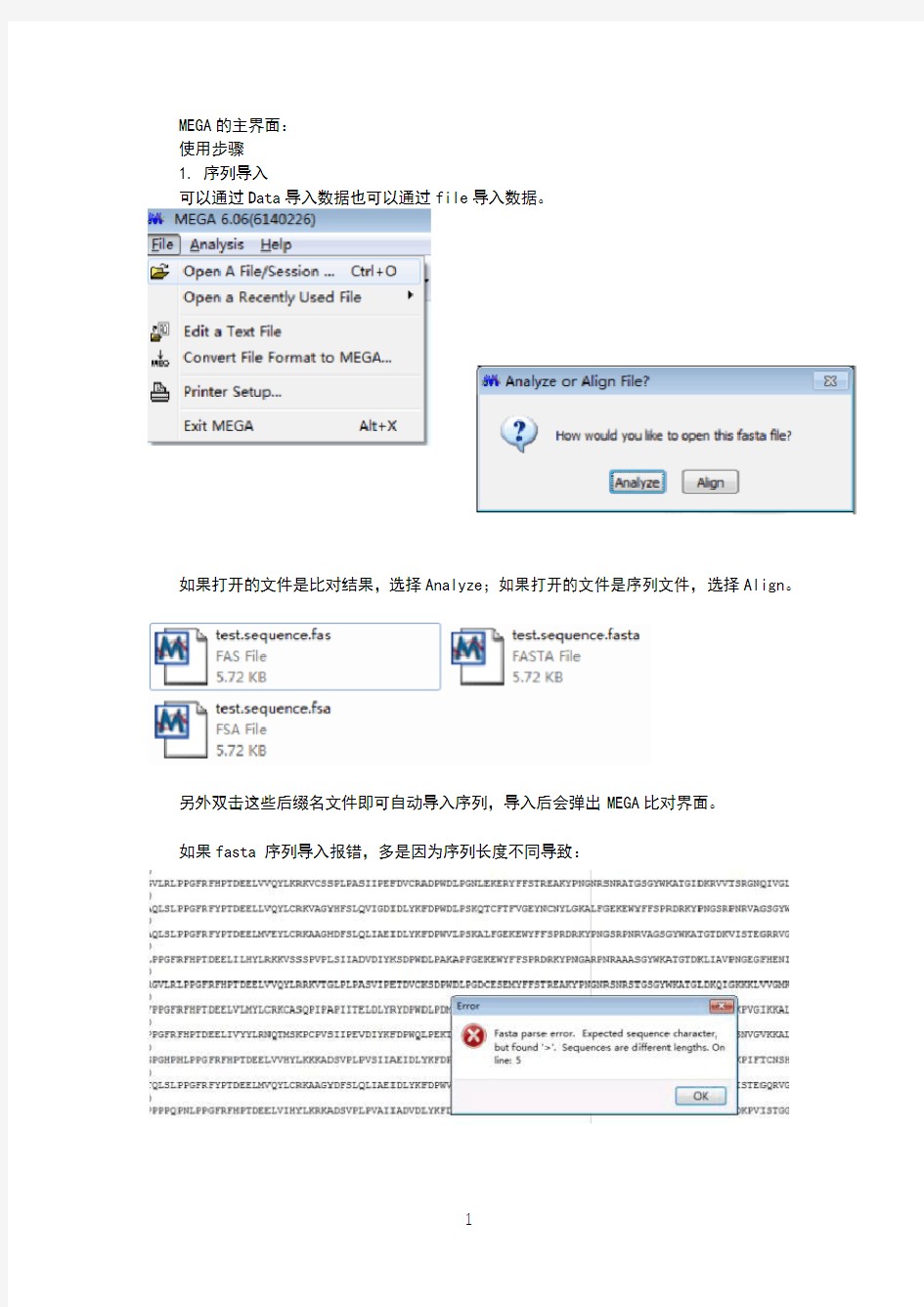

MEGA的主界面:
使用步骤
1. 序列导入
可以通过Data导入数据也可以通过file导入数据。
如果打开的文件是比对结果,选择Analyze;如果打开的文件是序列文件,选择Align。
另外双击这些后缀名文件即可自动导入序列,导入后会弹出MEGA比对界面。
如果fasta 序列导入报错,多是因为序列长度不同导致:
如果序列长度不同,可以采用新建文件,将序列文件导入的方法。
步骤:Align → Edit/Build Alignment → create a new alignment → Data → open → Retrieve sequences from File
将复制输入的序列另存输出看看。
步骤:data → Export alignment → fasta format
序列长度都被用横线补齐了。
2. 多序列比对
选择muscle或者clustalw进行比对:clustalw 一般用于DNA ,muscle多用于蛋白。
在比对之前需先选中要进行比对的序列(Shift),还可以对序列或者序列名进行编辑(双击)。
比对参数选择:
保存比对文件,进化树分析提供数据。
一般导出的比对结果保存为fasta格式,或者直接点击保存按钮将结果,保存为二进制的mas或meg文件。
3. 构建进化树
导入数据:将刚刚另存的meg 文件重新导入到mega程序中(直接拖入工作界面),并选择构建进化树。
参数选择:参数设置,Bootstrap method一般选择1000~1500;第一次绘图时建议选择500,这样运行速度会比价快,结果合适再调至1000重新进行进化分析。
描述:
进化树可视化:
View→Tree/Branch style选择树的模式,也可以通过右图菜单进行选择。
发散树
环状树
进化树简单美化:
可视化文件的保存:
?
Newick——标准树文件,用于下游可视化软件的导入
?
png——压缩图像文件
?
pdf——矢量图像文件
保存为png/pdf,显示不全怎么办
——将图和图注复制到WORD中保存即可(Image Copy to Clipboard 粘贴到WORD)。