
MEGA软件的使用
Mega软件输入数据的格式
Mega软件输入数据的格式比较简单,在众多遗传学分析软件中是比较容易制作的一种。
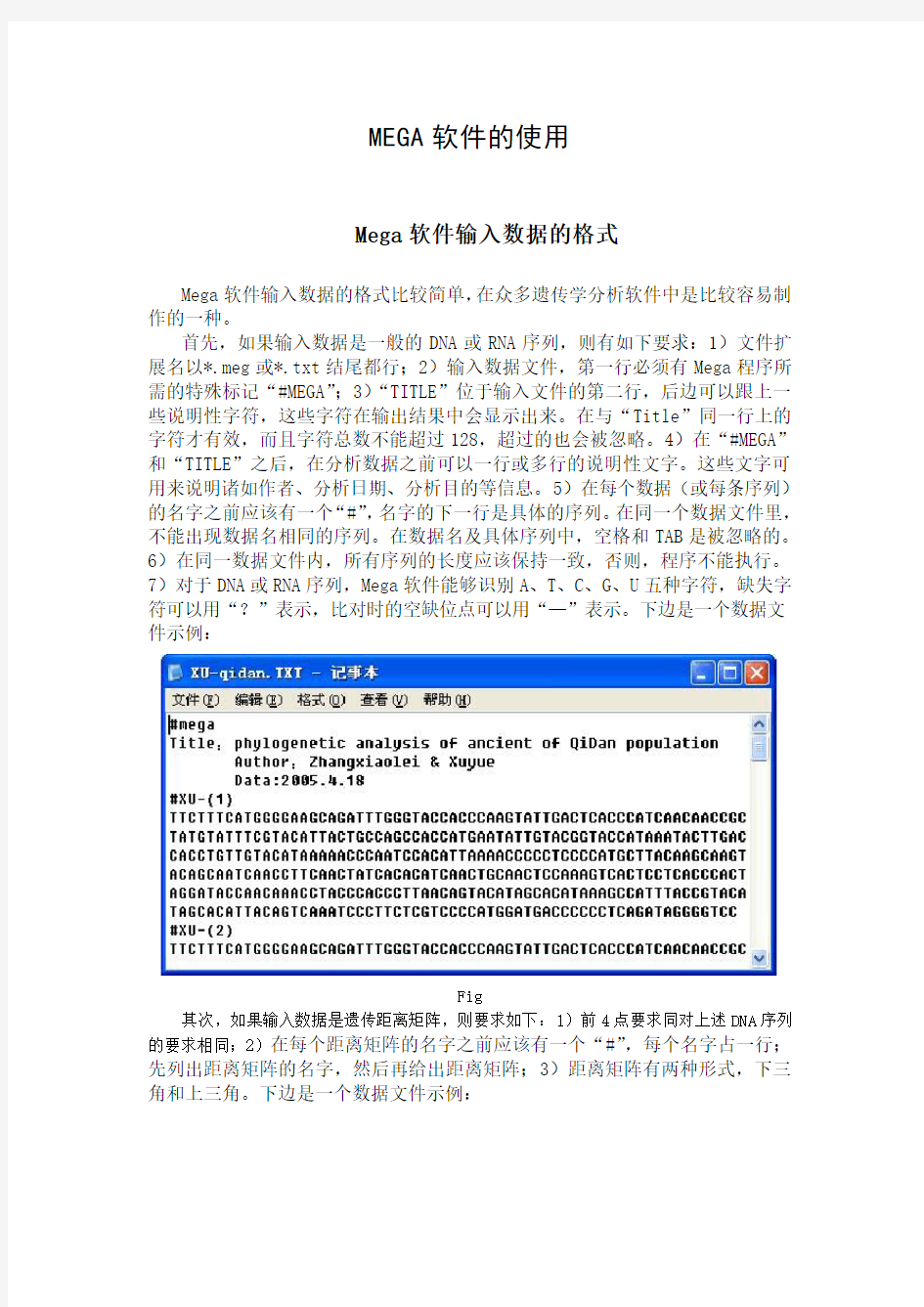
首先,如果输入数据是一般的DNA或RNA序列,则有如下要求:1)文件扩展名以*.meg或*.txt结尾都行;2)输入数据文件,第一行必须有Mega程序所需的特殊标记“#MEGA”;3)“TITLE”位于输入文件的第二行,后边可以跟上一些说明性字符,这些字符在输出结果中会显示出来。在与“Title”同一行上的字符才有效,而且字符总数不能超过128,超过的也会被忽略。4)在“#MEGA”和“TITLE”之后,在分析数据之前可以一行或多行的说明性文字。这些文字可用来说明诸如作者、分析日期、分析目的等信息。5)在每个数据(或每条序列)的名字之前应该有一个“#”,名字的下一行是具体的序列。在同一个数据文件里,不能出现数据名相同的序列。在数据名及具体序列中,空格和TAB是被忽略的。6)在同一数据文件内,所有序列的长度应该保持一致,否则,程序不能执行。7)对于DNA或RNA序列,Mega软件能够识别A、T、C、G、U五种字符,缺失字符可以用“?”表示,比对时的空缺位点可以用“—”表示。下边是一个数据文件示例:
Fig
其次,如果输入数据是遗传距离矩阵,则要求如下:1)前4点要求同对上述DNA序列的要求相同;2)在每个距离矩阵的名字之前应该有一个“#”,每个名字占一行;先列出距离矩阵的名字,然后再给出距离矩阵;3)距离矩阵有两种形式,下三角和上三角。下边是一个数据文件示例:
Fig
下图是距离矩阵的示意图,左边是下三角矩阵,右边是上三角矩阵。
Fig
再次,如果数据是测序图谱的形式,直接导入即可。下图是测序图谱示例:
Fig
MEGA界面及操作
Mega是一款操作十分简便的遗传学分析软件,其界面十分友好,即使初学者也很易上手。
1、数据的录入及编辑
Mega软件能够接受多种数据格式,如FASTA格式、Phylip格式、PAUP数据格式等等。而且Mega软件专门提供了把其他格式的数据转换位Mega数据格式的程序。
首先,打开Mega程序,有如下图所示的操作界面:
Fig
单击工具栏中的“File”按钮,会出现如下图所示的菜单:
Fig
从上图可以看出,下拉菜单有“Open Data”(打开数据)、“Reopen Data”(打开曾经打开的数据,一般会保留新近打开的几个数据)、“Close Data”(关闭数据)、“Export Data”(导出数据)、“Conver To MEGA Format”(将数据转化为MEGA格式)、“Text Editor”(数据文本编辑)、“Printer Setup”(启动打印)、“Exit”(退出MEGA程序)。单击“Open Data”选项,会弹出如下菜单:
Fig
浏览文件,选择要分析的数据打开,单击“打开”按钮,会弹出如下操作界面:
Fig
此程序操作界面,提供了三种选择数据选择:Nucleotide Sequences(核苷酸序列)、Protein Sequences(蛋白质序列)、Pairwise Distance(遗传距离矩阵)。根据输入数据的类型,选择一种,点击“OK”即可。如果选择“Pairwise Distance”,则操作界面有所不同;如下图所示:
Fig
根据遗传距离矩阵的类型,如果是下三角矩阵,选择“Lower Left Matrix”即可;如果是上三角矩阵,选择“Upper Right Matrix”即可。点击“OK”按钮,即可导入数据。如果是核苷酸数据,则读完之后,会弹出如下对话框:
Fig
如上图,如果是编码蛋白质的核苷酸序列,则选择“Yes”按钮;如果是不编码蛋白质的核苷酸序列,则点击“No”按钮。之后,会弹出如下操作窗口:
Fig
此作界面的名称是“Sequence Data Explorer”,在其最上方是工具栏“Data”、“Display”、“Highlight”等,然后是一些数据处理方式的快捷按钮,在操作界面的左下方是每个序列的名称。显示序列占了操作界面的绝大部分,与第一个序列相同的核苷酸用“.”表示,发生变异的序列则直接显示。
如果在弹出的对话框中,点击“OK”,即选择输入的数据是编码蛋白质的DNA 序列。那么会再弹出如下对话框:
Fig
此操作界面提供了多种生物的遗传密码方式的选择,如Vertebrate Mitochondrial(脊椎动物线粒体)、Invertebrate Mitochondrial(非脊椎动物线粒体)、Yeast Mitochondrial(酵母线粒体)等等。
点击此操作界面的“Add”按钮,可以添加密码子表格,其编辑界面如下图所示:
Fig
通过此操作界面可以创建、修改密码子表格。点击“OK”按钮可以返回“Select Genetic Code”操作界面。
点击“Select Genetic Code”操作界面的“Delect”按钮,可以删除一个密码子表。
点击“Select Genetic Code”操作界面的“Edit”按钮,可以对已经存在的密码子表格。其操作界面与“Genetic Code Table”相同。
点击“Select Genetic Code”操作界面的“View”按钮,可以浏览选中的密码子表格。
点击“Select Genetic Code”操作界面的“Statistics”按钮,可以统计密码子表格的一些信息,如每种密码子的频率、同义位点数、非同义位点数等。
点击点击“Select Genetic Code”操作界面的“OK”按钮,会弹出如上图所示的“Sequence Data Explorer”操作界面。如果点击“Cancel”按钮,也会弹出此操作界面,但是此时会把数据默认为非编码的DNA序列。
单击“Sequence Data Explorer”操作界面工具栏的“Data”按钮,有如下图所示的下拉菜单:
Fig
下拉菜单有六个选项:“Write Data To File”(将数据转到文件中,利用此选项可以把Mega数据格式的数据转化成其它格式)、“Translate/Untranslate”(是否翻译,这个选项只有所分析的DNA序列是编码序列时才被激活)、“Selcet Genetic Code Table”(选择遗传密码表,这个选项只有所分析的DNA序列是编码序列时才被激活)、“Setup/Selcet Genes&Domains”(选择或设置基因或结构域)、“Setup/Select Taxa&Group”(对数据进行分组)、“Quit Data Viewer”(退出此浏览框)。
单击“Write Data To File”选项,会弹出如下对话框:
Fig
Title框显示的内容是数据文件中“TITLE”之后的内容。Description框显示的内容是数据文件中对整体数据描述的内容。
Format选项提供一个下来菜单,通过此下拉菜单可以把数据转化为MEGA格式、Nexus(PAUP4.0)格式,PHYLIP3.0格式、Nexus(PAUP3.0/MacClade)格式。
Writing site numbers 选项也提供一个下拉菜单,通过此下来菜单可以把给每个核苷酸标序号,“None”为不显示序号,“For each site”为每个位点显示序号,“At the end of line”在每一行行末显示序号。
Missing Data and alignment gaps选项也提供了一个下拉式菜单,这个菜单包括:“Include sites with miss/ambiguous data gaps”(显示缺失位点及模糊位点以及空缺)、“Exclude sites with miss/ambiguous data gaps”(不显示缺失位点及模糊位点以及空缺)、“Exclude sites with miss/ambiguous data only”(仅不显示缺失位点及模糊位点)、“Exclude sites with alignment gaps only”(仅不显示比对是的空缺部分)。
如上述操作界面中的选项,点击“OK”按钮,会弹出如下界面:
Fig
此操作界面中的文字可以拷贝到文本文档中。如果在“Squence Data Explorer”操作界面的工具栏中选择“Highlight”中的“Varible sites”选项,则单击“Write Data To File”选项,会弹出如下对话框:
Fig
我们会发现与上述“Exporting Sequence Data”操作界面相比,在最下方增加了一个“Selceted sites to Include”下拉菜单框,此框包含:All sites (所有位点)、“Only highlighted sites”(只显示相互之间有变异的位点)、“Only unhighlighted sites”(只显示相互之间无变异的位点)三个选项。如上图中的操作界面中的选项,点击“OK”按钮,则会弹出如下对话框:
Fig
可以看出,在此操作界面中,仅显示了有变异的位点。这样的数据形式在转
化成“NetWork”遗传分析软件所需的数据格式时很方便。
单击“Sequence Data Explorer”操作界面的工具栏中“Data”中的“Setup/Selcet Genes&Domains”选项,会弹出如下对话框:
Fig
通过此操作界面可以检测、确定、选择结构域,为某些位点添加标签等。这个操作界面包括两大部分:“Define/Edit/Select”和“Site Labels”。通过操作界面中“Genes/Domain”的子菜单“Data”可以设置,起始位点和末位点。通过“Codon Start”选项,可以选择编码的起始位置。在操作界面下端有一排按钮:“Add Gene”、“Add Domain”、“Delete/Edit”、“Expand”。通过“Add Gene”按钮可以添加或插入一个新的基因,通过“Add Domain”按钮可以添加或插入一个新的结构域,通过“Delete/Edit”按钮可以对数据进行编辑和删除,通过“Expand”可以展开数据,或仅显示第一水平的数据。
点击“Site Labels”按钮,上述操作界面变为如下图所示:
Fig
点击上述操作界面中的“Close”按钮,返回“Sequence Data Explorer”操作界面。选择工具栏“Data”下拉菜单中的“Setup/Select Taxa&Groups”选项,弹出如下图所示操作界面:
Fig
如上图操作界面,点击“New Group”按钮可以创建一个新的组,点击“Delete Group”按钮可以删除一个已经存在的组,在操作界面的中间竖排有五个按钮,
同最上端两个按钮可以把数据移入或移出一个选定的组,点击第三个按钮可以对选定的组进行重新命名,点击“+”按钮可以创建一个新的组,点击“—”按钮可以删除一个已经存在的组。注意,组的名字不能与任何一个样本重名。点击“Close”按钮,“Sequence Data Explorer”操作界面。点击此操作界面中的“Display”按钮,会弹出如下操作菜单:
Fig
从上述操作界面图看,下拉菜单共有:“Show Only Selected Sequences”(仅显示选中的序列)、“Use Identical Symbol”(利用同一标记符号)、“Color Cells”(色彩单元)、“Sort Sequences”(序列分类)、“Restore Input Order”(恢复输入序列的顺序)、“Show Sequence Names”(显示序列名字)、“Show Group Names”(显示序列所在的组的名字)和“Change Font”(改变字体)八个选项。
选择“Show Only Selected Sequences”选项,只有被选中的序列才会在界面中显示,不过软件默认的是所有输入的序列都是被选中的,不过软件使用者是可以修改哪些序列被选中。
选择“Use Identical Symbol”选项,那么与第一个序列相同的核苷酸将用“.”显示,与之相比,发生变异的核苷酸才以“A、T、C、G”的形式显示。
选择“Color Cells”选项,不同的核苷酸将用不同的颜色显示,如下图所示。“Sort Sequences”选项有四个子选项:“By Sequence Name”(通过序列名字排列)、“By Group Name”(通过组的名字排列)、“By Group&Sequence Name”(通过组和序列的名字排列)、“As per Taxa&Group Organizer”()。
选择“Restore Input Order”选项,则序列排列顺序恢复到与输入数据文件中的顺序一样。
选择“Show Sequence Names”选项,则每个序列的名字被显示。选择“Show Group Names”,则每个序列所在的组的名字将被显示。
选择“Change Font”选项,可以改变序列名字、组名及其序列本身的字体大小及颜色,默认的字体大小是“小五”,默认的字体颜色是黑色,默认的字型
是常规,无下划线、删除线。
Fig
点击“Sequence Data Explorer”操作界面的“Highlight”选项,会有如下图所示的下拉菜单选项:
Fig
由上图可以看出,“Highlight”的下拉菜单共有七个选项:“Conserved Sites”(C,保守位点)、“Variable sites”(V,变异位点)、“Parsim-Info sites”(P,简约信息位点)、“Singleton sites”(S,单独位点)、“0-fold Degenerate sites”(0,未简并位点)、“2-fold Degenerate sites”(2,2倍简并位点)、“4- fold Degenerate sites”(4,4倍简并位点);其中后三个选项,只有在输入的序列是编码序码时才被激活。
选择“Conserved Sites”选项,所有的保守位点,即没有发生变异的位点,将被突出显示,位点的总数目将在状态栏(操作界面最下端)显示。
选择“Variable sites”选项,所有的变异位点,将被突出显示,位点的总数目将在状态栏(操作界面最下端)显示。
选择“Parsim-Info sites”选项,所有简约变异位点(即变异至少包括两种类型的核苷酸或氨基酸)将被突出显示,位点的总数目将在状态栏(操作界面最下端)显示。
选择“Singleton sites”选项,单突变(变异至少包括两种类型的核苷酸或氨基酸,而且在所有样本中仅发生一次)的位点,将被突出显示,位点的总数目将在状态栏(操作界面最下端)显示。
选择“0-fold Degenerate sites”选项,那些所有突变都是非同义突变的位点,将被突出显示,位点的总数目将在状态栏(操作界面最下端)显示。此选项只有在输入数据中含有编码蛋白的DNA序列时才被激活。
选择“2- fold Degenerate sites”选项,那些在所有突变中同义突变占1/3的位点,将被突出显示,位点的总数目将在状态栏(操作界面最下端)显示。此选项只有在输入数据中含有编码蛋白的DNA序列时才被激活。
选择“4- fold Degenerate sites”选项,那些所有突变全部是同义突变的位点,将被突出显示,位点的总数目将在状态栏(操作界面最下端)显示。此选项只有在输入数据中含有编码蛋白的DNA序列时才被激活。
点击“Sequence Data Explorer”操作界面的“Statistics”选项,会有如下图所示的下拉菜单选项:
Fig
从上图可以看出,此下拉菜单总共有六个选项:“Nucleotide Composition”(核苷酸组成)、“Nucleotide Pair Frequence”(核苷酸配对频率)、“Codon Usage”(密码子使用)、“Amino Acid Composition”(氨基酸组成)、“Use All Selected Sites”(利用所有选择的位点)、“Use Only Highlighted Sites”(仅利用突出显示的位点)。
选择“Nucleotide Composition”选项,可以计算得到,每条序列中A、T、C、G及U的百分含量,以及总的核苷酸个数,还可以得到整个数据中A、T、C、G及U的百分含量。如果数据是编码蛋白质的DNA序列,那么还可以得到每种核苷酸在密码子各个位置的比例。
选择“Nucleotide Pair Frequence”选项,可以计算DNA序列中核苷酸配对的频率。这个选项有两个子菜单:“Directional(16 Pairs)”和“Undirectional (10 Pairs)”。一个是有方向性的,一个是没有的。
选择“Codon Usage”选项,能够统计出每种密码子的使用频率。
选择“Amino Acid Composition”选项,能够统计出每条序列中各种氨基酸的组成百分含量,以及总的氨基酸个数。还可以计算出整个数据中每种氨基酸的组成百分含量。此选项只有在输入数据是氨基酸的条件下才被激活。
选择“Use All Selected Sites”选项,在计算统计时,可以利用所有被选中的位点。
选择“Use Only Highlighted Sites”选项,在计算分析时,仅利用那些被突出显示的位点进行计算。
在菜单栏的下方是一些常用的快捷方式,如下图示:
Fig
上图图标中,所对应的操作从左到右依次是:“Write Data To File”(将数据转到文件中)、“Setup/Select Taxa&Group”(对数据进行分组)、“Setup/Selcet Genes&Domains”(选择或设置基因或结构域)、“Use Identical Symbol”(利用同一标记符号)、“Color”(进行色彩设置)、“Conserved Sites”(C,保守位点)、“Variable sites”(V,变异位点)、“Parsim-Info sites”(P,简约信息位点)、“Singleton sites”(S,单独位点)、“0-fold Degenerate sites”(0,未简并位点)、“2-fold Degenerate sites”(2,2倍简并位点)、“4- fold Degenerate sites”(4,4倍简并位点)、将核苷酸序列翻译为蛋白质序列。
点击“Sequence Data Explorer”界面的“Data”下拉菜单中的“Quit Data Viewer”选项,即可关闭此操作界面,返回到Mega操作的主界面。
2、遗传距离的计算
2.1 遗传距离模型的选择
点击Mega操作主界面的“Distances”按钮,会弹出一个下拉菜单。如下图所示:
Fig
从上图易知,此菜单包括如下选项:“Choose Model”(选择模型,即选择计算遗传距离的模型)、“Compute Pairwise”(计算遗传配对差异)、“Compute Overall Mean”(计算包括所有样本在内的平均遗传距离)、“Compute With Group Means”(计算组内平均遗传距离)、“Compute Between Groups Means”(计算组间平均遗传距离)、“Compute Net Between Groups Means”(计算组间平均净遗传距离)、“Compute Sequence Diversity”(计算序列分歧度)。
“Compute Sequence Diversity”选项包括四个子菜单:“Mean Diversity Within Subpopulations”(亚群体内部平均序列多态性)、“Mean Diversity for Entire Population”(整个人群平均序列多态性)、“Mean Interpopulaional Diversity”(群体内部平均序列多态性)、“Coefficient of Differentiation”(遗传变异系数)。
点击“Choose Model”选项,会弹出如下操作界面:
Fig
从上述操作界面可以看出,通过此对话框可以选择计算遗传距离的模型等。
“Data Type”显示数据的类型:Nucleotide(Coding)(编码蛋白质的DNA 序列)、Nucleotide(不编码蛋白质的DNA序列)、Amino Acid(氨基酸序列)。
通过“Model”选项可以选择,计算遗传距离的距离模型。点击“Model”一行末端的按钮会弹出一选择栏。
Fig
如上图所示,对于非编码的核苷酸序列Mega程序提供了八种距离模型:“Number of Difference”(核苷酸差异数)、“P-distance”(P距离模型)、“Jukes-Cantor”(Jukes和Cantor距离模型)、“Kimura 2-Parameter”(Kimura 双参数模型)、“Tajima-Nei”(Tajima和Nei距离模型)、“Tamura 3-parameter”(Tamura 三参数模型)、“Tamura-Nei”(Tamura和Nei距离模型)、“LogDet (Tamura kumar)”(对数行列式距离模型)。
对于编码的核苷酸序列,其遗传距离模型如下图所示:
Fig
如上图所示,对于编码蛋白质的DNA序列,Mega程序提供了一下几种模型:“Nei-Gojobori Method”,“Modified Nei-Gojobori Methoed”、“Li-Wu-Luo Method”、“Pamilo-Bianchi-Li Method”、“Kumar Method”。其中Nei-Gojobori 方法和修正的Nei-Gojobori方法都包含三种距离模型:“Number of Differences”、“P-distance”、“Jukes-Cantor”。对于氨基酸序列,Mega所提供的遗传距离模型如下图所示:
Fig
如上图所示,对于氨基酸序列,Mega程序提供了一下六种遗传距离模型:“Number of Differences”(氨基酸差异数)、“P-distance”(P距离模型)、“Poisson Correction”(泊松校正距离模型)、“Equal Input”(等量输入距离模型)、“PAM Matrix(Dayhoff)”(PAM距离矩阵模型)、“JTT Matrix (Jones-Taylor-Thornton)”(JTT距离矩阵模型)。
在“Analysis Preference”操作界面中,“Pattern Among Lineages”仅提供了一个选项:“Same(Homogenous)”“,也就是说样本之间是有一定同源性的。“Rates among sites”提供了两个选项:“Uniform Rates”和“Different(Gamma Distributed)”。“Uniform Rates”意味着所有序列的所有位点的进化速率是相同的。选择“Different(Gamma Distributed)”,意味着序列位点之间的进化速率是不相同的,可以利用Gamma参数来校正,系统提供了四个数值可供选择:2.0、1.0、0.5、0.25;软件使用者也可以自行决定Gamma参数的大小。设置完毕后,在此界面中点击“OK”按钮,即可返回Mega操作主界面。
选择主操作界面“Distance”中的“Compute Pairwise”选项,可以计算样本之间的遗传距离的大小,其操作界面如下图所示: