AG-221 (Enasidenib)_突变体异柠檬酸脱氢酶2(IDH2)抑制剂_1446502-11-9_Apexbio
- 格式:pdf
- 大小:476.80 KB
- 文档页数:2

产品名: Pyridostatin 修订日期: 6/30/2016产品说明书化学性质产品名:Pyridostatin Cas No.:1085412-37-8 分子量:596.64 分子式:C31H32N8O5 别名:RR-82;RR82;RR 82 化学名:4-(2-aminoethoxy)-2-N,6-N-bis[4-(2-aminoethoxy)quinolin-2-yl]pyri dine-2,6-dicarboxamide SMILES:C1=CC=C2C(=C1)C(=CC(=N2)NC(=O)C3=CC(=CC(=N3)C(=O)NC4=NC5=CC=CC=C5C(=C4)OCCN)OCCN)OCCN 溶解性:>20.85mg/mL in DMSO 储存条件:Store at -20°C 一般建议: For obtaining a higher solubility , please warm the tube at 37°Cand shake it in the ultrasonic bath for a while.Stock solution can bestored below -20°C for several months.运输条件:Evaluation sample solution : ship with blue iceAll other available size: ship with RT , or blue ice upon request生物活性靶点 :Cell Cycle/Checkpoint 信号通路:G-quadruplex 产品描述:Pyridostatin 是合成的G-四联体小分子稳定剂[1]。
G-四联体是一种DNA 二级结构,通常存在于染色体或端粒的末端。
由于G-四联体也存在于一系列原癌基因(包括c-kit 、K-ras 和Bcl-2)的启动子中,因而被认为参与基因复制和转录的调节过程。


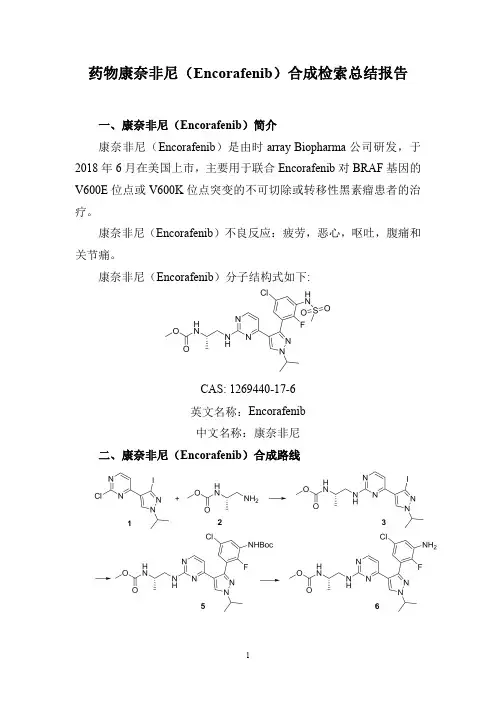
药物康奈非尼(Encorafenib)合成检索总结报告一、康奈非尼(Encorafenib)简介康奈非尼(Encorafenib)是由时array Biopharma公司研发,于2018年6月在美国上市,主要用于联合Encorafenib对BRAF基因的V600E位点或V600K位点突变的不可切除或转移性黑素瘤患者的治疗。
康奈非尼(Encorafenib)不良反应:疲劳,恶心,呕吐,腹痛和关节痛。
康奈非尼(Encorafenib)分子结构式如下:CAS:1269440-17-6英文名称:Encorafenib中文名称:康奈非尼二、康奈非尼(Encorafenib)合成路线三、康奈非尼(Encorafenib )合成检索总结报告(一)康奈非尼(Encorafenib )中间体3的合成合成方法实验步骤参考文献合成方法一Under nitrogen protection,Intermediate compound 1(0.6g,1.7mmol),Intermediate Compound 2(0.23g,1.7mmol),Sodium carbonate (0.71g,6.8mmol)was added in DMSO (20ml)solution,Warming up to 90°C,The reaction was stirred at this temperature for 16h,After the reaction is complete,DCM (30ml ×3)extraction,Combine the organic phase,Dry over anhydrous sodium sulfate,Concentrate for column separation (eluent:Petroleum ether /ethyl acetate (v /v)=1:2),The product 3was obtained in an amount of 0.65g,yield 84%.CN110256408;(2019);(A);CN110256408;(2019);(A)合成方法二A solution of Intermediate 2-chloro-4-(3-iodo-l-isopropyl--lH-pyrazol-4-yl)pyrimidine (1.4g,4.01mmol)1,(S)-methyl l-aminopropan-2-ylcarbamate 2(0.8g,6mmol)and triethylamine (2.8ml,20mmol)in isopropanol (30ml)and dioxane (20mL)was heated in a sealed vessel at 125o C for 48hours.The cooled mixture was concentrated under vacuum,and aqueous sodium bicarbonate was added to the residue.The mixture was extracted with ethyl acetate,and the combined extracts were dried over sodium sulfate,filtered,and concentrated.The crude residue was purified by silica gel columnchromatography (1:2hexanes/ethyl acetate eluant)to provide the title compound 3as a white solid.WO2011/25927;(2011);(A1)English(二)康奈非尼(Encorafenib )中间体5的合成。

New sialyl Lewis x mimic containing an a -substitutedb 3-amino acid spacerSilvana Pedatella,a,*Mauro De Nisco,a Beat Ernst,b Annalisa Guaragna,aBeatrice Wagner,b Robert J.Woods c and Giovanni Palumbo aa Dipartimento di Chimica Organica e Biochimica,Universita`di Napoli Federico II,Via Cynthia,4I-80126Napoli,Italy bInstitute of Molecular Pharmacy,Pharmacenter,University of Basel,Klingelberstrasse,50CH-4056Basel,Switzerland cComplex Carbohydrate Research Center,University of Georgia,315Riverbend Road,Athens,GA 30602,USAReceived 11June 2007;received in revised form 20August 2007;accepted 2October 2007Available online 7October 2007Abstract—A highly convergent and efficient synthesis of a new sialyl Lewis x (sLe x )mimic,which was predicted by computational studies to fulfil the spacial requirements for a selectin antagonist,has been developed.With a b 2,3-amino acid residue L -galactose (bioisostere of the L -fucose moiety present in the natural sLe x )and succinate are linked,leading to a mimic of sLe x that contains all the required pharmacophores,namely the 3-and 4-hydroxy group of L -fucose,the 4-and 6-hydroxy group of D -galactose and the carboxylic acid of N -acetylneuraminic acid.The key step of the synthesis involves a tandem reaction consisting of a N-deprotection and a suitable O !N intramolecular acyl migration reaction which is promoted by cerium ammonium nitrate (CAN).Finally,the new sialyl Lewis x mimic was biologically evaluated in a competitive binding assay.Ó2007Elsevier Ltd.All rights reserved.Keywords:sLe x ;Selectin antagonist;b 3-Amino acid;Glycomimetic1.IntroductionSelectins 1are involved in the orderly migration of leuco-cytes from blood vessels to the sites of inflammation.2Although extravasation of leucocytes represents an essential defense mechanism against inflammatory stim-uli,excessive infiltration of leucocytes into the surround-ing tissue can cause acute or chronic reactions as observed in reperfusion injuries,stroke,psoriasis,rheu-matoid arthritis,or respiratory diseases.2An early step in this inflammatory cascade is mediated by selectin/carbo-hydrate interactions.Data from both selectin knock out mice 3a–c and LAD type 2patients,3d clearly demon-strate that the selectin–carbohydrate interaction is a pre-requisite for the inflammatory cascade to take place.Since the tetrasaccharide sLe x (1,Fig.1)is the carbo-hydrate epitope recognized by E-selectin,4it became the lead structure for the design of selectin antagonists.5The search for novel selectin antagonist with enhanced adhesion and improved pharmacokinetic properties 5has led to numerous classes of antagonists.In the initial con-tributions,6,7the structure–activity relationship was eluci-dated,revealing the essential pharmacophores which are the carboxylic acid function of N -acetylneuraminic acid,the 3-and 4-hydroxyl group of L -fucose and the 4-and 6-hydroxyl group of D -galactose (highlighted in Fig.1).8In addition,it has been shown that the D -Glc-NAc moiety is not involved in binding.Its principal func-tion is that of a rigid spacer to accommodate the appropriate spacial orientation of L -fucose and D -galact-ose.5For the design of simplified sLe x antagonists 5a dual strategy was pursued by (1)eliminating monosaccharide moieties by non-carbohydrate linkers,and (2)substitut-ing metabolically labile O-glycosidic bonds.Considering the glycoaminoacid mimics reported by Wong,9which exhibit remarkable pharmacological0008-6215/$-see front matter Ó2007Elsevier Ltd.All rights reserved.doi:10.1016/j.carres.2007.10.001*Corresponding author.Tel.:+39081674118;fax:+39081674119;e-mail:pedatell@unina.itAvailable online at Carbohydrate Research 343(2008)31–38activity,we report herein the synthesis and the biological evaluation of the scaffold 2,a member of a new family of antagonists based on a dihydroxylated b -amino acid as a substitute of galactose.This scaffold is decorated with a suitably modified L -fucose and a carboxylic acid moiety mimicking the native N -acetylneuraminic acid.The corresponding target molecule 2(Fig.1)therefore contains (2S ,3S )-2-hydroxy-b 3-serine which is coupled with the free amino group of 6-amino-6-deoxy-L -galact-ose (L -Fuc replacement)and,at the other end,with a succinic acid (D -NeuNAc replacement).An important factor for affinity is the spacial orientation of the phar-macophores in the bioactive conformation.5d,e This is expected to be realized with the rigid diamide core.2.Results and discussionA conformational analysis of 2by molecular dynamic studies indicated that the low energy conformer-2pre-sented in Figure 1exhibits distances between the carbox-ylate and C-1and C-2of L -galactose (9.2and 9.1A˚,respectively),which are similar to the one found in the bioactive conformation 10of sLe x .Conformer-2is only less stable by 5.29kJ/mol compared to its lowest energy conformation and is characterized by differing flexibili-ties of the dihedral angles h ,u and x (Fig.2).Whereas h and u show considerable flexibility over the 10,000ps period of the MD simulation,x is rather stiff,leading to a partial preorganization of 2in the bioactive conformation.The synthesis started with the preparation of 6-amino-6-deoxy-L -galactose derivative 4from commercially available L -galactose according to a known procedure 11(Scheme 1).According to this procedure,the acetona-tion to achieve 1,2:3,4-di-O -isopropyliden-L -galactose(3),using CuSO 4/H 2SO 4in acetone,gave only poor yields (55%).Using an improved procedure 12(polystyryl diphenyl phosphine–iodine complex,prepared in situ,in anhy-drous acetone,at rt under nitrogen)3was obtained in 95%yield.Tosylation of the primary hydroxy group fol-lowed by substitution with sodium azide and catalytic hydrogenation (Pd/H 2)afforded the free amine 4in 69%overall yield from L -galactose.(2S ,3S )-2-Hydroxy-b 3-serine methyl ester derivative 7,was obtained diastereoselectively 13from commercially available O -benzyl-L -serine via enolate formation (by KHMDS)of the corresponding fully protected b 3-serine 5and the coupling of the enolate with racemic 2-[(4-methylphenyl)sulfonyl]-3-phenyloxaziridine (6)(Scheme 2).The hydrolysis of methyl ester 7afforded 8in excellent yield without any traces of racemization.Then,the 2-hydroxy-b 3-amino acid 8was coupled with the previously prepared 6-amino-L -galactose deriv-ative 4by treatment with DCC and HOBT in CH 2Cl 2,yielding 9in good yield (75%)(Scheme 3).The last step of the synthesis was the introduction of a carboxylate side chain at the N-terminus of 9.The first attempt to remove the 4-methoxybenzyl ether protection (PMB)with cerium ammonium nitrate (CAN)14was accompanied by the formation of several byproducts due to the cleavage of C-2–C-3bond of the b -aminoac-idic moiety.To avoid a possible interference of the free hydroxyl group with CAN,compound 9was first acetyl-ated and then treated with CAN.A single product was formed,which turned out not to be the desired N-depro-tected derivative,but the product of a subsequent O !N intramolecular acetyl migration.15When this O !N intramolecular acyl migration was applied to ester 10,which was obtained byesterificationFigure 1.Identical spacial orientation of the pharmacophores in sLe x (1)and the glycoaminoacid mimic 2.32S.Pedatella et al./Carbohydrate Research 343(2008)31–38of 9with 3-carbomethoxypropionyl chloride,product 11could be isolated in excellent 90%yield (Scheme 3).Finally,the removal of the protecting groups by hydrogenolysis on Pd/C in an ultrasound bath (!12),followed by alkaline hydrolysis (!13),and acidic hydrolysis (TFA/H 2O)of the isopropylidene groups (Scheme 3)yielded 2as an anomeric mixture (a /b =1:2).3.Biological evaluationVarious formats of cell-free competitive binding assays under static conditions have been reported.16–18We used microtiter plates coated with human recombinant E-selectin/IgG or P-selectin/IgG,which were incubated with 2and commercially available sLe x/biotin-polyacry-Figure 2.The plots A,B,and C show the fluctuations of the h ,u ,and x torsional angles in compound 2monitored during the 10,000ps MD simulation in a box of TIP3P water molecules;A,B:the h and u dihedral angles are highly variable within the MD simulation time frame;C:the x torsion angle remains practically unchanged during the MD simulation.S.Pedatella et al./Carbohydrate Research 343(2008)31–3833late.After unbound ligand and polymer were washed from the plate,streptavidin/horseradish peroxidase con-jugate was added to enable colorimetric determination of binding.18b As a reference compound,sialyl Lewis x with an IC50value of0.9mM for E-selectin and P3mM for P-selectin was used.With2,a moderate inhibition of the interaction of sLe x/E-selectin (IC50P6mM)could be detected.However,the interac-tion of P-selectin with the sLe x-polyacrylate polymer could not be inhibited with2(see Table1).4.ConclusionThe new sLe x mimic2was designed and synthesized starting from an L-galactose derivative4and orthogo-nally protected(2S,3S)-2-hydroxy-b3-serine8in six steps.Our synthesis features the use of cerium ammo-nium nitrate(CAN)to perform a useful O!N intra-molecular acyl migration reaction on the succinoyl ester10.However,biological testing revealed that this sLe x mimic shows only low affinity of E-selectin(IC50=6mM)and no activity(IC50>10mM)forP-selectin,respectively.Since the spatially correct arrangement of the phar-macophores can be realized in2,the low affinity can alsobe due to the considerable conformationalflexibility ofthe dihedral angles h and u(Fig.2A and B)leading tosubstantial cost of entropy upon binding.5.Experimental5.1.General methodsTriphenyl phosphine polymer-bound was purchasedfrom Fluka Chemical Co.All moisture-sensitive reac-tions were performed under a nitrogen atmosphere usingoven-dried glassware.Solvents were dried over standarddrying agents and freshly distilled prior to use.Reac-tions were monitored by TLC(precoated silica gel plateF254,Merck).Column chromatography:Merck Kiesel-gel60(70–230mesh);flash chromatography:MerckKieselgel60(230–400mesh).Optical rotations weremeasured at25±2°C in the stated solvent.1H and 13C NMR spectra were recorded on NMR spectro-meters operating at400or500MHz and50,100or125MHz,respectively.Wherever necessary,two-dimen-Table1.Biological evaluation dataTest compound E-Selectin(mM)P-Selectin(mM)Sialyl Lewis x(1)0.9P3Glycoaminoacid(2)P6>1034S.Pedatella et al./Carbohydrate Research343(2008)31–38sional1H–1H COSY experiments were carried out for complete signal bustion analyses were performed using CHNS analyzer.5.2.1,2:3,4-Di-O-isopropylidene-a-L-galactopyranose(3) To a magnetically stirred suspension of dry polystyryl diphenyl phosphine(1.12g,%3.34phosphine units)in anhydrous acetone(10mL)at rt,a solution of I2 (0.85g,3.34mmol)in the same solvent(30mL)was added dropwise in the dark and under dry nitrogen atmosphere.After15min,solid L-galactopyranose (0.33g,1.67mmol)was added in one portion to the sus-pension.TLC monitoring(CHCl3/CH3OH,9:1)showed that the starting sugar was completely consumed within 30min.The reaction mixture was thenfiltered through a glass sinter funnel and washed with acetone.The solvent was removed under reduced pressure and the solid resi-due recrystallized from CHCl3/hexane(1:2)to give thefinal product3(0.41g,95%yield);½a 25D À59.5(c1.5,CHCl3),lit.19½a 25D À55.0(c3.6,CHCl3).1H and13CNMR spectral data matched that reported.125.3.6-Amino-6-deoxy-1,2:3,4-di-O-isopropylidene-a-L-galactopyranose(4)The title compound was prepared following May’s pro-cedure11starting from3.5.4.(2S,3S)-4-(Benzyloxy)-3-[di(4-methoxybenzyl)-amino]-2-hydroxybutanoic acid(8)To a stirring solution of7,prepared as already re-ported,13(1.04g, 2.00mmol)in MeOH(13mL)was added dropwise aq NaOH(1.0M solution in H2O, 4.0mL)at0°C.The reaction,allowed to warm to rt, was stirred for6.5h and then it was diluted with EtOAc (100mL).The organic phase was treated with a solution of HCl(3M,2·100mL),washed with brine(50mL) and dried over Na2SO4.The solvent was removed under reduced pressure and the residue was purified byflash chromatography(CHCl3/MeOH,9:1)to afford8(0.90g,92%)as a pale yellow oil.½a 25D +44.0(c0.9,CHCl3);1H NMR(500MHz,CDCl3):d 3.45(ddd, 1H,J3,211.0,J3,4a9.6,J3,4b3.4Hz,H-3),3.82(s,6H, 2·OCH3), 3.84(d,1H,J2,311.0Hz,H-2), 3.89(d, 2H,J a,b12.0Hz,2·CH a PMB),3.98(dd,1H,J4a,4b 11.3,J4a,39.6Hz,H a-4),4.10(dd,1H,J4b,4a11.3,J4b,33.4Hz,H b-4),4.22(d,2H,J b,a12.0Hz,2·CH b PMB),4.59(d,1H,J a,b11.7Hz,CH a Ph),4.73(d,1H,J b,a 11.7Hz,CH b Ph), 6.89(d,4H,J ortho8.8Hz,ArH), 7.25(d,4H,J ortho8.8Hz,ArH),7.38–7.48(m,5H, PhH);13C NMR(125MHz,CDCl3)d54.2,55.1,60.1, 64.1,66.2,73.9,114.6,128.1,128.6,131.6,136.9, 160.2,175.0.Anal.Calcd for C27H31NO6:C,69.66;H, 6.71;N,3.01.Found:C,69.42;H,6.69;N,3.03.5.5.N1-(1,2:3,4-Di-O-isopropylidene-6-deoxy-a-L-galac-topyranos-6-yl)-(2S,3S)-4-(benzyloxy)-3-[di(4-methoxy-benzyl)amino]-2-hydroxybutanamide(9)HOBT(0.35g,2.58mmol)was added to a stirred solu-tion of b2,3-amino acid8(0.60g,1.29mmol)in anhy-drous CH2Cl2(10mL)at rt.After30min,to the resulting mixture a solution of compound4(0.30g, 1.17mmol)and DCC(0.40g,1.42mmol)in the same solvent(10mL)was added dropwise over5min at 0°C.The solution was allowed to warm slowly to rt and,after15h,most of the solvent was evaporated under reduced pressure and replaced by EtOAc.The precipitate wasfiltered and the solution was washed with saturated NaHCO3solution,brine until neutral, then dried(Na2SO4),and concentrated under reduced pressure.Chromatography of the crude residue on silica gel(hexane/EtOAc,1:1)afforded the oily coupling prod-uct9(0.62g,75%).½a 25D+8.5(c0.9,CHCl3);1H NMR (500MHz,CDCl3)d1.31,1.32,1.43,1.45(4s,12H, 4·CH3), 3.24(ddd,1H,J60a;60b13:9;J60a;508:1; J60a;NH4:4Hz,H a-60), 3.36–3.43(m,1H,H-3), 3.51 (ddd,1H,J60b;60a13:9;J60b;506:8;J60b;NH4:9Hz,H b-60), 3.67(d,2H,J a,b13.2Hz,2·CH a PMB),3.73(d,2H, J b,a13.2Hz,2·CH b PMB), 3.79(s,6H,2·OCH3),3.80–3.85(m,1H,H-50),3.92(dd,1H,J4a,4b10.7,J4a,34.4Hz,H a-4), 3.95–4.08(m,3H,H-40,H-2,H b-4), 4.27(dd,1H,J20;104:9;J20;301:9Hz,H-20), 4.50(dd, 1H,J30;407:8;J30;201:9Hz,H-30), 4.55(d,1H,J a,b 11.7Hz,CH a Ph),4.58(d,1H,J b,a11.7Hz,CH b Ph),5.48(d,1H,J10;204:9Hz,H-10),6.84(d,4H, J ortho8.3Hz,ArH),7.18(d,4H,J ortho8.3Hz,ArH), 7.28–7.40(m,5H,PhH),7.50–7.58(m,1H,NH);13C NMR(125MHz,CDCl3)d22.9,24.7,25.2,26.2,39.7, 54.7,55.5,59.5,66.3,68.4,69.6,70.7,71.0,71.7,73.7, 96.5,108.9,109.6,114.1,127.9,128.7,130.6,131.1, 138.1,159.0,173.8.Anal.Calcd for C39H50N2O10:C, 66.27;H,7.13;N,3.96.Found:C,66.50;H,7.11;N, 3.93.5.6.1-{[(1S,2S)-3-(Benzyloxy)-2-[di(4-methoxybenzyl)-amino]-1-[(1,2:3,4-di-O-isopropylidene-6-deoxy-a-L-galactopyranos-6-yl)carbamoyl]propyl}4-methyl succi-nate(10)3-Carbomethoxy propionyl chloride(0.17g,1.16mmol) and anhydrous pyridine(94l L,1.16mmol)were added to a solution of compound9in anhydrous CH2Cl2 (11mL)at0°C.The stirring solution was allowed to warm slowly to rt and after4h most of the solvent was evaporated under reduced pressure and replaced by EtOAc.The organic phase was washed with brine (2·30mL),dried(Na2SO4),and concentrated under re-duced pressure.The residue oil was purified by silica gel chromatography(hexane/EtOAc,8:2!6:4)to give theoily product10(0.56g,90%).½a 25D+18.0(c0.8,CHCl3);S.Pedatella et al./Carbohydrate Research343(2008)31–38351H NMR(500MHz,CDCl3)d1.29,1.32,1.43,1.45(4s, 12H,4·CH3),2.58–2.68(m,4H,CH2–CH2),3.05(ddd, 1H,J60a;60b13:9;J60a;508:8;J60a;NH3:5Hz,H a-60),3.51–3.56(m,1H,H-2), 3.60(d,2H,J a,b13.1Hz, 2·CH a PMB),3.65(d,2H,J b,a13.1Hz,2·CH b PMB), 3.66(s,3H,CH3OCO),3.67–3.80(m,10H,H b-60,H-50, 2·OCH3,2·H-3),4.13(dd,1H,J40;307:8;J40;501:6Hz, H-40),4.27(dd,1H,J20;104:9;J20;302:5Hz,H-20),4.44 (s,2H,CH2Ph), 4.56(dd,1H,J30;407:8;J30;202:5Hz, H-30),5.46(d,1H,J10;204:9Hz,H-10),5.51(d,1H,J1,2 4.3Hz,H-1),6.54(dd,1H,J NH;60b8:0;J NH;60a3:5Hz, NH),6.82(d,4H,J ortho8.7Hz,ArH),7.27(d,4H,J ortho 8.7Hz,ArH),7.30–7.39(m,5H,PhH);13C NMR (100MHz,CDCl3)d24.8,25.4,26.1,26.4,29.2, 29.6,39.6,52.2,54.4,55.6,58.3,67.2,67.8,71.0, 71.2,71.9,72.9,73.3,96.6,109.2,109.8,114.0, 127.9,128.1,128.7,130.5,132.1,138.7,159.0, 169.4,171.1,173.1.Anal.Calcd for C44H56N2O13:C, 64.38;H,6.88;N,3.41.Found:C,64.19;H,6.85;N, 3.43.5.7.Methyl3-{[(1S,2S)-1-[(benzyloxy)methyl]-2-hydroxy-2-([1,2:3,4-di-O-isopropylidene-6-deoxy-a-L-galactopyranos-6-yl]carbamoyl)ethyl]carbamoyl}pro-panoate(11)A solution of CAN(4.9mL,2.9mmol)in H2O was added to a stirring solution of compound10(0.48g, 0.58mmol)in MeCN at0°C.The reaction was kept to0°C for1h and then was allowed to warm to rt.After 18h,the reaction mixture was quenched by the addition of a saturated NaHCO3solution(50mL)and extracted with CHCl3.The organic layer was washed with brine until neutral,dried(Na2SO4),and concentrated under reduced pressure.The residue,after chromatography on silica gel(CHCl3/MeOH,95:5),gave the oily product11(0.20g,90%).½a 25D À11.0(c0.6,CHCl3);1H NMR(500MHz,CDCl3)d1.31,1.33,1.45,1.47(4s,12H, 4·CH3),2.50(t,2H,J1,26.8Hz,CH2–CH2),2.65(t, 2H,J2,1 6.8Hz,CH2–CH2), 3.28(ddd,1H, J60a;60b13:9;J60a;508:8;J60a;NH4:0Hz,H a-60),3.61–3.68 (m,5H,H b-60,CH a PMB,CH3OCO), 3.84–3.90 (m,2H,H-50,CH b PMB), 4.16(dd,1H, J40;307:9;J40;501:8Hz,H-40),4.23(br s,1H,H-1),4.30 (dd,1H,J20;104:9;J20;302:4Hz,H-20), 4.35–4.40(m, 1H,H-2), 4.48(d,1H,J a,b11.7Hz,CH a Ph), 4.52 (d,1H,J b,a11.7Hz,CH b Ph), 4.59(dd,1H, J30;407:9;J30;202:4Hz,H-30),5.51(d,1H,J10;204:9Hz, H-10),6.31(br d,1H,J NH,16.8Hz,NH serine),7.22–7.26(m,1H,NH galactosamine),7.30–7.40(m,5H,PhH); 13C NMR(100MHz,CDCl3)d24.8,25.3,26.3,26.4, 29.6,31.2,40.0,52.2,53.7,66.6,69.7,70.9,71.2,72.1, 73.5,96.7,109.1,109.9,128.2,128.4,128.9,137.8, 171.9,173.3,173.6.Anal.Calcd for C28H40N2O11:C, 57.92;H,6.94;N,4.82.Found:C,58.14;H,6.91;N, 4.84.5.8.Methyl3-{[(1S,2S)-2-hydroxy-1-(hydroxymethyl)-2-([1,2:3,4-di-O-isopropylidene-6-deoxy-a-L-galactopyr-anos-6-yl]carbamoyl)ethyl]carbamoyl}propanoate(12)A solution of compound11(0.20g,0.35mmol)in MeOH(4mL)was added to a stirring suspension of 5%palladium on carbon(0.07g)in the same solvent (5mL)and then was hydrogenated(1atm)at40°C. Theflask was immersed in an ultrasound cleaning bath filled with water and sonicated for22h.Then the sus-pension wasfiltered through CeliteÒand the solid washed twice with MeOH(2·5mL).The organic phase was evaporated down under reduced pressure to afford the oily product12(0.16g,93%).½a 25D+0.4(c0.5, CHCl3);1H NMR(500MHz,CDCl3)d 1.33, 1.36, 1.47,1.49(4s,12H,4·CH3),2.51–2.57(m,2H,CH2–CH2), 2.63–2.69(m,2H,CH2–CH2), 3.37(ddd,1H, J60a;60b13:4;J60a;508:3;J60a;NH4:4Hz,H a-60),3.65–3.72 (m,4H,H b-60,CH3OCO), 3.74(dd,1H,J Ha,Hb 11.9,J Ha,1 4.9Hz,CH a OH), 3.87(ddd,1H, J50;60a8:3;J50;60b3:9;J50;401:9Hz,H-50),4.05(dd,1H, J Hb,Ha11.9,J Hb,11.5Hz,CH b OH),4.15–4.18(m,1H, H-1),4.19(dd,1H,J40;307:8;J40;501:9Hz,H-40),4.27–4.30(m,1H,H-2),4.31(dd,1H,J20;104:9;J20;302:4Hz, H-20),4.61(dd,1H,J30;407:8;J30;202:4Hz,H-30),5.20 (br s,1H,OH),5.50(d,1H,J10;204:9Hz,H-10),5.90 (br s,1H,OH),6.77(d,1H,J NH,16.3Hz,NH serine), 7.54–7.64(m,1H,NH galactosamine);13C NMR (50MHz,CDCl3)d22.7,23.2,24.3,27.4,28.0,28.7, 38.1,50.2,54.0,60.5,64.4,68.8,69.1,70.1,75.5,94.6, 107.1,107.9,171.6,173.2,173.4.Anal.Calcd for C21H34N2O11:C,51.42;H,6.99;N,5.71.Found:C, 51.28;H,6.96;N,5.73.5.9.3-{[(1S,2S)-2-Hydroxy-1-(hydroxymethyl)-2-([1,2:3,4-di-O-isopropylidene-6-deoxy-a-L-galactopyr-anos-6-yl]carbamoyl)ethyl]carbamoyl}propanoic acid(13) One molar solution of aq NaOH(0.78mL,0.78mmol) was added to a stirring solution of compound12 (0.13g,0.26mmol)in MeOH(13mL)at0°C.The reac-tion mixture was allowed to warm slowly to rt and after 3h(TLC monitoring;CHCl3/CH3OH,8:2)was quenched with some drops of acetic acid until neutral. The precipitate wasfiltered and the organic phase was evaporated under reduced pressure to afford the oilyproduct13(0.11g,91%).½a 25D+9.0(c0.8,CH3OH); 1H NMR(400MHz,CD3OD)d1.33,1.36,1.43,1.47 (4s,12H,4·CH3), 2.40–2.52(m,4H,CH2–CH2), 3.35(dd,1H,J60a;60b14:0;J60a;508:1Hz,H a-60), 3.48 (dd,1H,J60b;60a14:0;J60b;504:4Hz,H b-60), 3.61–3.66 (m,2H,CH2OH),3.94–3.99(m,1H,H-50),4.20(br d,1H,J2,1 3.7Hz,H-2), 4.24–4.31(m,2H,H-40, H-1),4.34(dd,1H,J20;105:0;J20;302:5Hz,H-20),4.63 (dd,1H,J30;408:0;J30;202:5Hz,H-30), 5.48(d,1H, J10;205:0Hz,H-10);13C NMR(125MHz,CD3OD)36S.Pedatella et al./Carbohydrate Research343(2008)31–38d23.2,24.5,25.1,26.3,34.0,34.6,40.7,55.3,61.1,67.4,71.9,72.2,72.9,73.2,97.8,109.9,110.5,174.7,176.3,181.0.Anal.Calcd for C20H32N2O11:C,50.42;H,6.77;N,5.88.Found:C,50.28;H,6.79;N,5.89.5.10.3-{[(1S,2S)-2-Hydroxy-1-(hydroxymethyl)-2-([6-deoxy-L-galactopyranos-6-yl]carbamoyl)ethyl]carbam-oyl}propanoic acid(2)A solution of glycoaminoacid13(0.11g,0.23mmol)inTFA/H2O(9:1,5mL)was stirred at rt.After3h,thereaction solvent was evaporated under reduced pressureto afford a crude that was purified by Sephadex G-10column leading to the mimic2as a mixture of b and aanomers in2:1ratio(0.067g,80%).1H NMR(500MHz,D2O)d 2.33–2.41(m,4H,CH2–CH2),3.30–3.38(m,2.66H,H-20b,2·H-60b,2·H-60a),3.52(dd,0.66H,J30;2010:0;J30;403:3Hz,H-30b), 3.53–3.60 (m,2H,CH2OH), 3.63(br dd,0.66H,J50;60a7:4;J50;60b5:0Hz,H-50b), 3.68(dd,0.34H,J20;3010:3; J20;103:6Hz,H-20a), 3.72(br dd,0.34H,J30;2010:3; J30;403:1Hz,H-30a), 3.76(br d,0.66H,J40;303:3Hz, H-40b),3.82(br d,0.34H,J40;303:1Hz,H-40a),3.96–4.02(m,0.34H,H-50a),4.11–4.23(m,2H,H-1,H-2),4.43(d,0.66H,J10;207:8Hz,H-10b),5.12(d,0.34H, J10;203:6Hz,H-10a);13C NMR(125MHz,D2O)d 31.5,32.5,41.7(C-60b),41.8(C-60a),55.7(C-2),61.8(CH2OH),70.5(C-20a,C-50a),71.3(C-30a),71.4(C-40b),71.8(C-40a),73.6(C-1),74.1(C-20b),74.9(C-30b,C-50b),94.6(C-10a),98.8(C-10b).Anal.Calcd forC14H24N2O11:C,42.43;H,6.10;N,7.07.Found:C,42.30;H,6.09;N,7.09.putational methodsMolecular dynamics simulations were performed usingthe AMBER7.0suite programs20using the SANDER mod-ule.The simulated structure2was built up by module XLEAP of AMBER using the AMBER forcefield PARM99.21 The GLYCAM2000parameters22,23were implemented forthe oligosaccharide simulations.The charges were takenfrom ab initio calculations performed by GAUSSIAN98molecular modelling package,24using the Hartree–Fockmethod with6-31G*basic set.The MD simulation of2were performed with a time step of1fs in a cubic box ofwater8.0A˚to the side containing1102TIP3P watermolecules.25After heating and equilibration for50psthe simulations were performed for10ns,under periodicboundary conditions,at constant pressure(1atm),andconstant temperature(300K).Energy minimizationsand MD simulations were performed with a dielectricconstant of unity,and a cut-offvalue for non-bondedinteractions of8A˚.The1–4electrostatic and van derWaals interactions were scaled by the standard values(SCEE=1.2,SCNB=2.0).Post-processing of the trajectories(torsional,distance and energy analysis)was performed using the CARNAL module of AMBER7.0package.AcknowledgementsS.P.is grateful to Professor Steve Hanessian,who intro-duced her to the fascinating sLe x researchfield.We thank Oto Miedico for collecting valuable results while performing his master thesis work and Professor Romu-aldo Caputo for useful discussions.We are grateful to Dr Cristina de Castro for her help in the purification protocol of thefinal product.1H and13C NMR spectra were performed at Centro Interdipartimentale di Met-odologie Chimico-Fisiche(CIMCF),Universita‘di Na-poli Federico II.Varian Inova500MHz spectrometer is the property of Naples Laboratory of Consorzio Inter-universitario Nazionale La Chimica per l’Ambiente (INCA).References1.Kansas,G.S.Blood1996,88,3259–3287.2.(a)Lobb,R.R.In Adhesion.Its Role in InflammatoryDisease;Harlan,J.M.,Liu,D.Y.,Eds.;Freeman,W.H.: New York,1992,Chapter1,pp1–18;(b)Paulson,J.C.In Adhesion.It’s Role in Inflammatory Disease;Harlan,J.M., Liu, D.Y.,Eds.;Freeman,W.H.:New York,1992, Chapter2,pp19–42;(c)Varki,A.Proc.Natl.Acad.Sci.U.S.A.1994,91,7390–7397.3.(a)Tedder,T.F.;Steeber,D.A.;Chen,A.;Engel,P.FASEB J.1995,9,866–873;(b)McEver,R.;Moore,K.;Cummings,R.D.J.Biol.Chem.1995,270,11025–11028;(c)Lasky,L.Annu.Rev.Biochem.1995,64,113–139;(d)von Adrian,U.H.;Berger, E.M.;Ramezani,L.;Chambers,J.D.;Ochs,H.D.;Harlan,J.M.;Paulson,J.C.;Etzioni,A.;Arfors,K.-E.J.Clin.Invest.1993,91,2893–2897.4.(a)Phillips,M.;Nudelman,E.;Gaeta,F.C.;Perez,M.;Singhal,A.K.;Hakomori,S.-L.;Paulson,J.C.Science 1990,250,1130–1132;(b)Walz,G.;Aruffo,A.;Kolanus, W.;Bevilacqua,M.;Seed,B.Science1990,250,1132–1135;(c)Berg,E.L.;Robinson,M.K.;Mansson,O.;Butcher,E.C.;Magnani,J.L.J.Biol.Chem.1991,266, 14869–14872.5.For reviews see:(a)Ernst,B.;Kolb,H.C.;Schwardt,O.In The Organic Chemistry of Sugars;Levy,D.E.,Fu¨gedi, P.,Eds.;Taylor&Francis Group,2006;pp803–861;(b) Kaila,N.;Thomas,B.E.,IV Med.Res.Rev.2002,22, 566–601;(c)Simanek,E.E.;McGarvey,G.J.;Jablonow-ski,J.A.;Wong,C.-H.Chem.Rev.1998,98,833–862;(d) Kolb,H.C.;Ernst,B.Chem.Eur.J.1997,3,1571–1578;(e)Kolb,H.C.;Ernst,B.Pure Appl.Chem.1997,69,1879–1884;(f)Bertozzi,C.R.Chem.Biol.1995,2,703–708;(g)Musser,J.H.;Anderson,M.B.;Levy,D.E.Curr.Pharm.Des.1995,1,221–232;(h)Giannis,A.Angew.Chem.,Int.Ed.Engl.1994,33,178–180.6.Brandley, B.K.;Kiso,M.;Abbas,S.;Nikrad,P.;Srivasatava,O.;Foxall, C.;Oda,Y.;Hasegawa, A.Glycobiology1993,3,633–641.S.Pedatella et al./Carbohydrate Research343(2008)31–38377.Ramphal,J.Y.;Zheng,Z.-L.;Perez,C.;Walker,L.E.;DeFrees,S.A.;Gaeta,F.C.A.J.Med.Chem.1994,37, 3459–3463,and literatures cited herein.8.(a)Stahl,W.;Sprengard,U.;Kretschmar,G.;Kunz,H.Angew.Chem.,Int.Ed.Engl.1994,33,2096–2098;(b) Somers,W.S.;Tang,J.;Shaw,G.D.;Camphausen,R.T.Cell2000,103,467–479.9.Cappi,M.W.;Moree,W.J.;Qiao,L.;Marron,T.G.;Weitz-Schmidt,G.;Wong, C.-H.Bioorg.Med.Chem.1997,5,283–296.10.(a)Scheffler,K.;Ernst,B.;Katopodis,A.;Magnani,J.L.;Wang,W.T.;Weisemann,R.;Peters,T.Angew.Chem., Int.Ed.Engl.1995,34,1841–1844;(b)Scheffler,K.;Brisson,J.-R.;Weisemann,R.;Magnani,J.L.;Wong,W.T.;Ernst,B.;Peters,T.J.Biomol.NMR1997,9,423–436.11.May,J.A.,Jr.;Sartorelli,J.J.Med.Chem.1979,22,971–976.12.Pedatella,S.;Guaragna,A.;D’Alonzo,D.;De Nisco,M.;Palumbo,G.Synthesis2006,2,305–308.13.Caputo,R.;Cecere,G.;Guaragna, A.;Palumbo,G.;Pedatella,.Chem.2002,17,3050–3054.14.Reetz,M.T.;Ro¨hrig,D.Angew.Chem.,Int.Ed.Engl.1989,28,1706–1709.15.Sohma,Y.;Hayashi,Y.;Skwarczynski,M.;Hamada,Y.;Sasaki,M.;Kimura,T.;Kiso,Y.Biopolymers(Peptide Sci.)2004,76,344–356.16.Rao,B.N.;Anderson,M.B.;Musser,J.H.;Gilbert,J.H.;Schaefer,M.E.;Foxall,C.;Brandley,B.K.J.Biol.Chem.1994,269,19663–19666.17.(a)Foxall,C.;Watson,S.R.;Dowbenko,D.;Fennie,C.;Lasky,L.A.;Kiso,M.;Hasegawa,A.;Asa,D.;Brandley,B.K.J.Cell.Biol.1992,117,895–902;(b)Jacob,G.S.;Kirmaier,C.;Abbas,S.Z.;Howard,S.C.;Steininger,C.N.;Welply,J.K.;Scudder,P.Biochemistry1995,34, 1210–1217;(c)Galustian,C.;Childs,R.A.;Yuen,C.-T.;Hasegawa,A.;Kiso,M.;Lubineau,A.;Shaw,G.;Feizi,T.Biochemistry1997,36,5260–5266;(d)Marinier, A.;Martel, A.;Banville,J.;Bachand, C.;Remillard,R.;Lapointe,P.;Turmel,B.;Menard,M.;Harte,W.E.,Jr.;Wright,J.J.K.;Todderud,G.;Tramposch,K.M.;Bajorath,J.;Hollenbaugh,D.;Aruffo,A.J.Med.Chem.1997,40,3234–3237.18.(a)Weitz-Schmidt,G.;Stokmaier,D.;Scheel,G.;Nifan-t’ev,N.E.;Tuzikov,A.B.;Bovin,N.V.Anal.Biochem.1996,238,184–190;(b)Thoma,G.;Magnani,J.L.;O¨hrlein,R.;Ernst,B.;Schwarzenbach,F.;Duthaler,R.O.J.Am.Chem.Soc.1997,119,7414–7415.19.Schmidt,O.T.Meth.Carbohydr.Chem.1969,1140–1142.20.Case,D.A.;Pearlman,D.A.;Caldwell,J.W.;Cheatham,T. E.,III;Wang,J.;Ross,W.S.;Simmerling, C.L.;Darden,T.A.;Merz,K.M.;Stanton,R.V.;Cheng,A.L.;Vincent,J.J.;Crowley,M.;Tsui,V.;Gohlke,H.;Radmer, R.J.;Duan,Y.;Pitera,J.;Massova,I.;Seibel,G.L.;Singh,U.C.;Weiner,P.K.;Kollman,P.A.AMBER7;University of California:San Francisco,2002.21.Cornell,W.D.;Cieplak,P.;Bayly,C.I.;Gould,I.R.;Merz,K.M.;Ferguson,D.M.;Spellmeyer,D.C.;Fox, T.;Caldwell,J.W.;Kollman,P.A.J.Am.Chem.Soc.1995,117,5179–5197.22.Kirschner,K.N.;Woods,R.J.Proc.Natl.Acad.Sci.U.S.A.2001,98,10541–10545.23.Woods,R.J.;Dwek,R.A.;Edge,C.J.;Fraser-Reid,B.J.Phys.Chem.1995,99,3832–3846.24.Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;Scuseria,G.E.;Robb,M.A.;Cheeseman,J.R.;Zakrzewski,V.G.;Montgomery,J.A.,Jr.;Stratmann,R.E.;Burant,J.C.;Dapprich,S.;Millam,J.M.;Daniels,A.D.;Kudin,K.N.;Strain,M.C.;Farkas,O.;Tomasi,J.;Barone,V.;Cossi, M.;Cammi,R.;Mennucci,B.;Pomelli,C.;Adamo,C.;Clifford,S.;Ochterski,J.;Petersson,G.A.;Ayala,P.Y.;Cui,Q.;Morokuma,K.;Malick,D.K.;Rabuck,A.D.;Raghavachari,K.;Foresman,J.B.;Cioslowski,J.;Ortiz, J.V.;Stefanov,B.B.;Liu,G.;Liashenko,A.;Piskorz,P.;Komaromi,I.;Gomperts,R.;Martin,R.L.;Fox,D.J.;Keith,T.;Al-Laham,M.A.;Peng,C.Y.;Nanayakkara,A.;Gonzalez, C.;Challacombe,M.;Gill,P.M.W.;Johnson, B.;Chen,W.;Wong,M.W.;Andres,J.L.;Head-Gordon,M.;Replogle,E.S.;Pople,J.A.GAUSSIAN 98,9ed.;Gaussian,1998.25.Jorgensen,W.L.;Chandrasekhar,J.;Madura,J. D.;Impey,R.W.;Klein,M.L.J.Chem.Phys.1983,79,926–935.38S.Pedatella et al./Carbohydrate Research343(2008)31–38。

Labetuzumab govitecan 是一种抗体药物偶联物(ADC),由以下三个主要部分组成:
1. 抗体Labetuzumab:这是一种靶向CEACAM5(癌胚抗原相关细胞粘附分子5)的单克隆抗体。
2. SN-38:这是拓扑异构酶I抑制剂的一种活性形式,它是伊立替康(irinotecan)的一个更活跃的代谢产物。
3. pH敏感连接子:这种连接子能够在肿瘤细胞内部的低pH环境中断开,从而释放SN-38,对肿瘤细胞产生毒性作用。
Labetuzumab govitecan的作用机制包括CEACAM5拮抗剂和TOP1抑制剂的双重作用。
它通过抗体Labetuzumab特异性地结合到CEACAM5阳性的肿瘤细胞上,然后利用SN-38的强大细胞毒性来杀死这些肿瘤细胞。
总的来说,这种ADC的设计旨在提高化疗药物的靶向性,减少对正常细胞的毒副作用,从而提高治疗效果和患者的耐受性。
Labetuzumab govitecan的研发和应用是肿瘤治疗领域的一个重要进展,特别是在针对CEACAM5表达较高的肿瘤类型中。





Overcoming Clopidogrel Resistance:Discovery of Vicagrel as a Highly Potent and Orally Bioavailable Antiplatelet AgentJiaqi Shan,†,‡Boyu Zhang,†,‡Yaoqiu Zhu,§Bo Jiao,∥Weiyi Zheng,⊥Xiaowei Qi,#Yanchun Gong,†,‡Fang Yuan,#Fusheng Lv,#and Hongbin Sun *,†,‡†State Key Laboratory of Natural Medicines and ‡Center of Drug Discovery,College of Pharmacy,China Pharmaceutical University,24Tongjia Xiang,Nanjing 210009,China §MetabQuest Research and Consulting,1720Oak Avenue,Suite 403,Evanston,Illinois 60201,United States ∥Department of Pharmacology,School of Pharmacy,Shandong University,44Wenhua West Road,Jinan 250012,China ⊥Concord Pharmatech Co.,Ltd.,9Xinglong Road,Nanjing 211800,China #Jiangsu Vcare PharmaTech Co.,Ltd.,15Wanshou Road,Nanjing 211800,China*Supporting Information metabolism studies,since its potency was comparable to that of Preliminary pharmacokinetic study results showed that the bioavailability than that generated from clopidogrel,implying a much lower In summary,9a (vicagrel)holds great promise as a more potent advantages over clopidogrel:(1)no drug resistance for CYP2C19much lower effective dose;(3)faster onset of action.Clopidogrel (1)(Figure 1)is an oral antiplatelet agent used to prevent blood clots in coronary artery disease,peripheral vascular disease,and cerebrovascular disease.Currently,dual treatment with aspirin and clopidogrel is the cornerstone of antiplatelet therapy for patients with acute coronary syndrome (ACS)and prevention of thrombotic events after percutaneous coronary intervention (PCI)with stenting.1The drug works byirreversibly inhibiting P2Y12receptor,a subtype of adenosine diphosphate (ADP)receptors on the platelet membrane.It is a prodrug requiring two-step metabolic conversion by the cytochrome P450(CYP)system to generate clopidogrel active metabolite (AM,5)via clopidogrel thiolactone (4a )(Scheme 1).2However,nonresponsiveness or poor responsiveness to clopidogrel therapy occurs in up to 30%of Caucasian patients (∼2%are poor metabolizers (PMs),∼26%are intermediate metabolizers (IMs)),3especially in PMs carrying CYP2C192*loss-of-function polymorphism,leading to lower levels of the active metabolite of clopidogrel,less inhibition of platelets,and a 1-to 5-fold higher risk for death,myocardial infarction,and stroke in comparison with noncarriers.4,5This refers to the concept of clopidogrel resistance (CR).In 2010,the FDA put a blackbox warning on clopidogrel to make patients and healthcare professionals aware that CYP2C19PMs are at high risk of treatment failure.Currently,clinical practice to overcome CR includes (1)the adjunctive use of cilostazol,(2)increasing the dose of clopidogrel,(3)the use of new antiplatelet agents,such as prasugrel (2)and ticagrelor (3)(Figure 1).6However,the above approaches may increase the risk of major and fatal bleedings and reduce patient compliance.7Taken together,there are still unmet medical needs for the treatment of ACS and its complications,especiallyReceived:January 10,2012Published:March 19,2012Figure 1.Structures of P2Y12receptor antagonists.the clinical management of CR.Novel antiplatelet agents with fast onset of action,less interindividual variability,and low risk of bleeding are urgently needed.Given the fact that clopidogrel is among the most widely prescribed drugs worldwide and its long-term safety profile hasbeen established after14years of clinical use,new drug discovery based on this old drug would be highly desirable.As Nobel laureate James Black said,“The most fruitful basis for the discovery of a new drug is to start with an old drug.”We envisioned that,like the metabolic pattern of prasugrel,ester prodrugs9might be readily converted to clopidogrel thiolactone(4a)by esterase-mediated hydrolysis and sub-sequently to clopidogrel AM(5)through only one CYP-dependent step(Scheme1).In this regard,9could be ideal drug candidates for overcoming CR without increasing the risk of bleeding and other side effects associated with the new antiplatelet agents.Herein,we report the identification of a series of2-hydroxytetrahydrothienopyridine derivatives as novel antiplatelet agents.We also describe the detailed structure−activity relationships(SARs)of this series of compounds.Preliminary efficacy,toxicity,and pharmacokinetic studies led to the discovery of vicagrel(9a)as a highly potent and orally bioavailable antiplatelet agent.■RESULTS AND DISCUSSIONChemistry.Optically pure(R)-methyl2-(2-substitued-phenyl)-2-[(4-nitrobenzenesulfonyl)oxy]acetates7a−c were prepared via reaction of mandelic acid and2-substituted mandelic acids with4-nitrobenzenesulfonyl chloride in the presence of triethylamine at low temperature(Scheme2).N-Alkylation of5,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2(4H)-one hydrochloride(8)with7a−c in the presence of potassium bicarbonate afforded thiolactones4a−c.Reaction of4a−c with acyl anhydrides,acyl chlorides,chlorocarbonic acid esters,or N-substituted carbamic chlorides in the presence of triethylamine or sodium hydride gave optically active2-hydroxytetrahydro-thienopyridine derivatives9a−v with(S)-configuration.The enantiomer of9a((R)-9a)and the racemic mixture of9a ((R,S)-9a)were prepared starting from optically pure(S)-methyl2-(2-chlorophenyl)-2-[(4-nitrobenzenesulfonyl)oxy]-acetate and racemic methyl2-(2-chlorophenyl)-2-[(4-nitrobenzenesulfonyl)oxy]acetate,respectively.The optical purity(%ee)of compounds9a−v and(R)-9a was determinedby chiral HPLC.Inhibition of ADP-Induced Platelet Aggregation inRats and SAR Analysis.Twenty-five2-hydroxytetrahydro-thienopyridine derivatives were evaluated for their inhibitoryeffect on ADP-induced platelet aggregation in rats at dose of3mg/kg.Clopidogrel(1)and prasugrel(2)were used as positivecontrols.ADP-induced platelet aggregation was determined byBorn’s method.8The assay results are summarized in Table1.In our preliminary tests,although clopidogrel showed strongpotency at a dose of10mg/kg(data not shown),it was almostinactive at a dose of3mg/kg.Prasugrel was the most potentone among this set of test pound9a exhibitedvery strong inhibitory effect on platelet aggregation and wasonly slightly less potent than prasugrel.2-Chloro substitutionseemed to be critical for the potency(e.g.,9a−c,l−o),sinceboth2-hydrogen and2-fluoro substitution resulted insignificant loss of potency(e.g.,9u,9v).2-Hydroxy estermoiety of the tetrahydrothienopyridine ring also had asignificant impact on potency.Interestingly,when the size of2-hydroxy alkyl esters increased,the potency decreasedaccordingly(e.g.,potency in the order9a>9b>9c>9d>9e),suggesting that the hindrance at the2-ester groups mightreduce their hydrolysis rates and thus result in lowerconcentrations of clopidogrel AM.The same tendency wasalso observed with carbonic acid esters(e.g.,potency,9m>9n>9p>9q).It was notable that the potency of carbonic acidester9m was similar to that of alkyl ester9a,indicating thatcarbonic acid esters could also be converted to clopidogrel AMas well as alkyl esters.Not surprisingly,clopidogrel thiolactone(4a)was a potent antiplatelet agent,albeit less potent than its Scheme1.Metabolic Activation of Clopidogrel and ProdrugDesignScheme2.Synthesis of Optically Active2-Hydroxytetrahydrothienopyridine Derivatives9a−v aa Reagents and conditions:(a)4-nitrobenzenesulfonyl chloride,CH2Cl2,Et3N,DMAP(cat.),0°C;(b)KHCO3,CH3CN,rt;(c)(R2CO)2O,CH3CN,Et3N,0°C to rt,or R2COCl,THF,Et3N orNaH.For R1and R2,see Table1.acetic ester 9a .Aromatic acid esters (e.g.,9f ,9g ,9h ,9i )were not as potent as alkyl esters,except for nicotinate ester 9l which exhibited strong potency.Substituted carbamate esters were inactive in this assay (e.g.,9s ,9t ).Preliminary tests had shown that 9a seemed to be the most promising drug candidate among this series of compounds,and thus,its enantiomer ((R )-9a )and racemic mixture ((R ,S )-9a )were synthesized and biologically evaluated in order to investigate the effect of configuration on potency.In contrast to 9a ,(R )-9a was almost inactive,while (R ,S )-9a had very weak activity.On the basis of the above results,9a was selected for further in vitro and in vivo metabolism studies.In Vitro Metabolic Activation Studies on Clopidogrel and 9a in Rat Liver Microsomes (RLMs).Clopidogrel (1)could be metabolically activated to form its active metabolite (AM,5)via the thiolactone intermediate (4a )under in vitro incubation conditions with liver microsomes 9or cDNA-expressed P450isozymes 10(Figure 2).The active metabolite (5)is chemically labile probably because of the reactive thiol function.Although it was reported that 5might be stable at 4°C for 24h in quenched incubation mixtures,10for theconvenience of LC −MS/MS-based qualitative and quantitative analyses as well as further purification for NMR studies,5was often derivatized in the incubation mixture with a variety of reagents that were reactive toward the thiol function including glutathione (GSH),acrylonitrile,3′-methoxyphenacyl bromide,N -ethylmaleimide,and dimedone.10,11In this study,glutathione was used to derivatize 5to form the active metabolite −glutathione disulfide adducts (AMGS,Figure 2).To test the metabolic activation of 9a and potential formation of the active metabolite (5),rat liver microsomal incubation was conducted for 9a in parallel with clopidogrel in the presence of NADPH and glutathione.After quenching and centrifugation,the supernatant was analyzed by LC −MS/MS.Incubation of clopidogrel with rat liver microsomes in the presence of GSH leads to NADPH-dependent formation of metabolites (Figure 3A)that exhibited a molecular ion at m /z of 661and a product ion spectrum that is in complete agreement with the formation of the clopidogrel AMGS disulfide adducts.LC −MS/MS analysis of the supernatant resulting from incubation of 9a with rat liver microsomes in the presence of GSH reveals NADPH-dependent formation of glutathione adducts at the same LCTable 1.Inhibitory Effect of 2-Hydroxytetrahydrothienopyridine Derivatives on ADP-Induced Platelet Aggregation in Rats at a Dose of 3mg/kgacompd R 1R 2%ee d platelet aggregation (%)1b99.073.7±5.22c 32.1±9.3**4a 98.148.6±14.6**9a Cl methyl 99.134.6±13.5**9b Cl ethyl 96.546.7±15.4**9c Cl n -propyl 96.352.8±7.7**9d Cl tert -butyl 99.163.4±16.2*9e Cl tert -amyl 99.570.0±23.09f Cl phenyl93.560.1±11.9**9g Cl 4-NO 2-phenyl 10075.6±7.09h Cl 4-MeO-phenyl 96.982.6±9.19i Cl 2-AcO-phenyl 96.077.9±4.99j Cl benzyl 93.553.8±10.4**9k Cl styryl98.784.7±8.79l Cl pyridine-3-yl 97.745.1±9.0**9m Cl methoxy 97.038.6±9.1**9n Cl ethoxy 97.353.0±11.6**9o Cl isopropoxy 97.550.2±9.3**9p Cl isobutoxy 95.565.6±23.29q Cl benzyloxy93.772.2±10.59r Cl phenoxymethyl 89.075.4±4.59s Cl dimethylamino 95.567.7±17.79t Cl pyrrolidine-1-yl 95.780.9±9.29u H methyl 72.081.1±6.09v F methyl86.962.9±12.8*(R )-9a 98.768.4±19.6(R ,S )-9a 63.7±8.0*vehicle75.7±7.7aAssay details are described in Experimental Section.Aggregation data refer to ex vivo measurements 2h after oral administration.(∗)P <0.05.(∗∗)P <0.01vs vehicle.Data are the mean ±SD,n =10.b Clopidogrel hydrogen sulfate was used as a positive control.c Prasugrel free base was used as a positive control.d ee refers to enantiomeric excess.Figure2.Proposed pathways of active metabolite and active metabolite−glutathione adduct formation via in vitro metabolic activation of clopidogrel and9a upon incubation with RLM in the presence of GSH and NADPH.Figure3.LC−MS-selective ion monitoring chromatogram(SIM,M+H+=661)of active metabolite−glutathione(AMGS−1/2/3/4)adducts in RLM incubation of clopidogrel(A)or9a(B)for30min.retention time as the AMGS adducts formed in clopidogrel incubation (Figure 3,B).The molecular ion (MS)spectrum (Figure 4,top)and product ion (MS2)spectrum (Figure 4,bottom)of the glutathione adducts detected in rat liver microsomal incubation of 9a were almost identical to those of the AMGS adducts in clopidogrel incubation (data not shown).The MS spectra of the glutathione adducts show the typical isotopic pattern that is in accordance with the monochloro-containing nature of the AMGS adducts.Fragmentation analysis (Figure 4,inserted)based on the exclusive product ions resulting from collision-induced dissociation (CID)of the molecular ion [M +H]+of m /z 661(MS2spectrum)confirms the formation of the AMGS adducts with the presence of both moieties of 9a and glutathione as well as the disulfide bond formation.The metabolic activation of 9a or clopidogrel confers on the structure of the active metabolite two additional stereochemical sites:one chiral center adjacent to the thio function and one geometric center of the ethylenic double bond,which provide the basis for the formation of four diastereomers.12Chromatographic separation resolved the four diastereomeric AMGS adducts formed from 9a into four peaks with retention times of 9.41min (AMGS −1),9.84min (AMGS −2),10.02min (AMGS −3),and 10.63min (AMGS −4)(Figure 3B).On the basis of the LC −MS/MS studies of in vitro rat liver microsomal incubation of 9a (see Supporting Information)paralleled to clopidogrel,the metabolic activation pathways of 9a are proposed in Figure 2.As shown in the first step,the acetate ester function of 9a undergoes hydrolysis probably by esterases presented in rat liver microsomes and gets deacetylated.The formed 2-hydroxytetrahydrothienopyr-idine (10)is a tautomer of thiolactone 4a ,which is the metabolic intermediate resulting from the first oxidative activation of clopidogrel (1).The metabolic activation pathways of clopidogrel and 9a converge after the first steps through this 2-hydroxytetrahydrothienopyridine −thiolactone tautomerization and share the subsequent pathways including the second step of NAPDH-dependent oxidative ring-opening of 4a that eventually lead to active metabolite formation (Figure 2).Pharmacokinetic Parameters of Clopidogrel Thiolac-tone after Oral Administration of Clopidogrel or 9a to Rats.The dose of clopidogrel or 9a at 24μmol kg −1was orally administered to SD male rats.Blood was collected at 0h (before dosing)and 0.25,0.5,1,2,4,6,8,24h postdose.The dose of clopidogrel thiolactone at 8μmol kg −1was intravenous administration to SD male rats to determine the conversion rate or bioavailability of clopidogrel or 9a to clopidogrel thiolactone.Blood was collected at 0h (before dosing)and 0.083,0.167,0.5,1,2,4,6,8,24h postdose.After sample cleanup,the plasma samples were subjected to LC −MS/MS analysis to determine the plasma concentrations of clopidogrel thiolactone (Figure 5).After oral dosing of clopidogrel,the C max ,T max ,t 1/2,and AUC 0−∞of clopidogrel thiolactone in plasma were 6.93±3.36μg L −1,0.583±0.382h,2.48±0.466h,and 32.2±10.9μg ·h L −1,respectively.After oral dosing of 9a ,the C max ,T max ,t 1/2,and AUC 0−∞of clopidogrel thiolactone in plasma were 67.2±42.3μg L −1,1.17±0.764h,2.19±1.68h,and 211±119μg ·h L −1,respectively.The aforementioned data proved that after oral administration,9a could be readily converted into clopidogrel thiolactone,and bioavailabilityofFigure 4.Averaged molecular ion (MS,top)spectrum and product ion (MS2,bottom)spectrum of the glutathione (AMGS)adducts detected in rat liver microsomal incubation of 9a in the presence of NADPH and glutathione.The inset is proposed fragmentation pathways of the molecular ion [M +H]+of AMGS at m /z of 661.clopidogrel thiolactone generated from 9a was 6-fold higher than that generated from clopidogrel at the same dose.Single-Dose Toxicity of 9a.Single-dose toxicity studies on 9a were performed in GLP laboratories.No mouse (n =10males,10females)died after receiving 5000mg/kg 9a by oral gavage in the 14-day observation period.Liver injury was observed,and no adverse effects were observed in other organs.After oral administration of 9a to beagle dogs,the no-observed-adverse-effect level (NOAEL)was 300mg/kg and the maximal tolerance dose (MTD)was higher than 2000mg/kg.The above data indicate that 9a has very low acute toxicity.■CONCLUSIONSAlthough in 2010,the FDA issued a “boxed warning ”of reduced effectiveness of clopidogrel in patients carrying CYP2C19loss-of-function alleles,there is still debate on whether there is a significant association between CYP2C19genotype and cardiovascular events in patients receiving clopidogrel.5,13,14Nevertheless,it seems no doubt that CYP2C19loss-of-function alleles lead to clopidogrel resistance.In this regard,the cardiovascular risk associated with clopidogrel resistance would be definitely an unnegligible factor,especially for severe ACS patients with PCI,since platelet-mediated thrombotic events might be amplified in the presence of both plaque disruption and interventional procedures.15On the other hand,most of the study population involved in the large-scale clinical trials on clopidogrel were Caucasians among whom the percent of CYP2C19poor metabolizers is only about 2%.By contrast,the percent of CYP2C19poor metabolizers in East Asians is much higher at 15−23%.16That is,the meta-analysis results 13,14based on those clinical trials may not truly reflect the risk associated with clopidogrel resistance,especially for East Asian rge-scale clinical trials addressing clopidogrel resistance are warranted to define the risk of major adverse cardiovascular outcomes among CYP2C19poor metabolizers treated with clopidogrel.In the present study,prodrug design based on clopidogrel thiolactone metabolite was mainly aimed at overcoming clopidogrel resistance.Therefore,252-hydroxytetrahydrothie-nopyridine derivatives were synthesized and evaluated for their inhibitory effect on ADP-induced platelet aggregation in rats.The animal study results showed that some compounds (e.g.,9a ,9b ,9j ,9l ,9m ,9n ,9o )exhibited potent activity as antiplatelet pound 9a was selected for further studies,since its potency was comparable with that of prasugreland was much higher than that of clopidogrel.In vitro metabolism studies revealed that,like clopidogrel,9a could be readily converted to clopidogrel active metabolite by rat liver microsomes.Preliminary pharmacokinetic study results showed that the bioavailability of clopidogrel thiolactone generated from 9a was 6-fold higher than that generated from clopidogrel,implying a much lower clinically effective dose and thus lower dose-related toxicity for 9a in comparison with clopidogrel.In summary,9a holds great promise as a more potent and a safer antiplatelet agent that might have the following advantages over clopidogrel:(1)no drug resistance for CYP2C19poor metabolizers;(2)lower dose-related toxicity due to a much lower effective dose;(3)faster onset of action due to its metabolic activation mechanism.From our point of view,the bleeding risk of 9a should be manageable,since under a proper dosing range,the efficacy of 9a would be equal to that of clopidogrel,and thus,in theory,the bleeding risk of 9a should not be higher than that of clopidogrel.Further preclinical trials on 9a (vicagrel)are currently being conducted in our laboratories to prove the above predictions.■EXPERIMENTAL SECTIONChemistry.Materials and General Methods.All commercially available solvents and reagents were used without further purification.Melting points were determined with a Bu c hi capillary apparatus and were not corrected.1H and 13C NMR spectra were recorded on an ACF *300Q Bruker or ACF *500Q Bruker spectrometer in CDCl 3,with Me 4Si as the internal reference,or in DMSO-d 6.Low-and high-resolution mass spectra (LRMS and HRMS)were recorded in electron impact mode.Reactions were monitored by TLC on silica gel 60F254plates (Qingdao Ocean Chemical Company,China).Column chromatography was carried out on silica gel (200−300mesh,Qingdao Ocean Chemical Company,China).The purity of all final compounds was determined to be ≥95%by analytical HPLC (equipment:Agilent 1100system with a VWD G1314A UV detector;column,Chiralpak IC,4.6mm ×250mm).Detailed analytical HPLC conditions for each final compound are described in the following section.(R )-M e t h y l 2-(2-C h l o r o p h e n y l )-2-(4-nitrophenylsulfonyloxy)acetate (7a).To a stirred mixture of (R )-2-hydroxy-2-(2-chlorophenyl)acetate (98.4g,0.49mol,99.0%ee)and Et 3N (91mL,0.65mol)in CH 2Cl 2(500mL)at 0°C was slowly added a solution of 4-nitrobenzenesulfonyl chloride (120g,0.54mol)in CH 2Cl 2(500mL).After being stirred for 4h at the same temperature,the mixture was quenched with water.The organic layer was separated,dried over anhydrous sodium sulfate,and concentrated under reduced pressure.The residue was recrystallized in MeOH to afford the title compound as a light yellow solid (154.5g,82.0%yield),98.1%ee (Chiral HPLC analytical conditions:Chiralpak IC,4.6mm ×250mm,eluting with 50%n -hexane +50%i -PrOH,flow rate 0.5mL/min,oven temperature 25°C,detection UV 220nm).1H NMR (300MHz,CDCl 3):δ8.30(d,2H,J =8.9Hz),8.07(d,2H,J =8.9Hz),7.39−7.21(m,4H),6.39(s,1H),3.57(s,3H).ESI-MS m /z 408.0[M +Na]+.(S )-Methyl 2-(2-Chlorophenyl)-2-(2-oxo-7,7a-dihydrothieno-[3,2-c ]pyridin-5(2H ,4H ,6H )-yl)acetate (4a).To a solution of (R )-methyl 2-(2-chlorophenyl)-2-(4-nitrophenylsulfonyloxy)acetate (58.1g,0.15mol)in CH 3CN (500mL)were added 5,6,7,7a-tetrahydrothieno[3,2-c ]pyridin-2(4H )-one hydrochloride (32.3g,0.17mol)and potassium bicarbonate (37.8g,0.38mol).After being stirred at room temperature for 26h under N 2atmosphere,the mixture was filtered and the liquid was concentrated under reduced pressure.The residue was purified by column chromatography to give a yellow oil,which was recrystallized in EtOH to afford the title compound as a white solid (35.4g,70%yield),mp 146−148°C,98.1%ee (determined after conversion to 9a ).[α]D19+114.0°(c 0.5,MeOH).1H NMR (300MHz,CDCl 3):δ7.54−7.51(m,1H),7.44−Figure 5.Plasma concentrations of clopidogrel thiolactone after oral administration of clopidogrel or 9a to SD rats at a dose of 24μmol kg −1.7.41(m,1H),7.32−7.26(m,2H),6.02(s,1H),4.91(s,1H),4.16 (m,1H),3.92(m,1H),3.73(s,3H),3.25(d,1H,J=12.3Hz),3.03 (d,1H,J=12.7Hz),2.65−2.59(m,1H),2.37−2.31(m,1H),1.93−1.79(m,1H).13C NMR(75MHz,CDCl3):δ198.4,170.7,167.1, 134.8,132.8,130.0,129.7,127.1,126.7,67.2,52.2,51.5,51.0,49.6, 33.8.ESI-MS m/z338.1[M+H]+.HRMS calcd for C16H17NO3SCl [M+H]+m/z338.0618,found338.0626.(S)-Methyl2-(2-Acetoxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)acetate(9a).To a stirred mixture of 4a(6.5g,19mmol,97.5%ee)and Et3N(5.4mL,38.5mmol)in CH3CN(100mL)at0°C was slowly added acetic anhydride(3.6mL, 38.5mmol).The mixture was stirred for2h at room temperature and quenched with water.Then AcOEt(100mL)was added to the mixture and the organic layer was washed with saturated sodium bicarbonate solution,brine and dried over anhydrous sodium sulfate. The organic layer was concentrated under reduced pressure and the residue was purified by column chromatography to afford a light yellow oil,which was recrystallized in EtOH to afford the title compound as a white solid(6.8g,93.2%yield),mp73−75°C,99.1% ee(Chiral HPLC analytical conditions:Chiralpak IC,4.6mm×250 mm,eluting with92%n-hexane+8%THF+0.1%Et2NH,flow rate 0.5mL/min,oven temperature25°C,detection UV254nm).[α]D23 +45.00°(c1.0,MeOH).1H NMR(300MHz,CDCl3):δ7.70−7.24 (m,4H),6.26(s,1H),4.92(s,1H),3.72(s,3H),3.60(ABq,2H,J =14.2Hz),2.90(s,2H),2.79−2.65(m,2H),2.26(s,3H).13C NMR (75MHz,CDCl3):δ170.7,167.2,149.1,134.2,133.3,129.4,129.3, 128.9,128.8,126.6,125.3,111.5,67.3,51.6,49.8,47.6,24.5,20.2.ESI-MS m/z380.0[M+H]+.HRMS calcd for C18H19NO4SCl[M+H]+ m/z380.0723,found380.0737.(S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl Propionate(9b).Following a procedure similar to that described for the preparation of9a except that an equivalent amount of propionic andydride was used in place of acetic anhydride,the title compound was obtained as a colorless oil in 66.0%yield,96.5%ee(Chiral HPLC analytical conditions:Chiralpak IC,4.6mm×250mm,eluting with90%n-hexane+10%i-PrOH+ 0.1%Et2NH,flow rate0.5mL/min,oven temperature25°C, detection UV254nm).[α]D20+36.00°(c0.50,MeOH).1H NMR(300 MHz,CDCl3):δ7.69−7.26(m,4H),6.26(s,1H),4.91(s,1H), 3.72(s,3H),3.59(ABq,2H,J=14.2Hz),2.88−2.87(m,2H), 2.78−2.76(m,2H),2.55(q,2H,J=7.7Hz),1.23(t,3H,J=7.4 Hz).13C NMR(75MHz,CDCl3):δ171.2,149.8,123.7,130.0,129.8, 129.5,129.1,127.2,125.6,111.7,106.2,67.8,52.2,50.3,48.2,27.4, 25.0,21.1,8.8.ESI-MS m/z394.1[M+H]+.HRMS calcd for C19H21NO4SCl[M+H]+m/z394.0883,found394.0880.(S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl Butyrate(9c).Following a procedure similar to that described for the preparation of9a except that an equivalent amount of butyric anhydride was used in place of acetic anhydride,the title compound was obtained in41.0%yield, 96.3%ee(Chiral HPLC analytical conditions:same as those for9b). [α]D20+32.00°(c0.50,MeOH).1H NMR(300MHz,CDCl3):δ7.69−7.24(m,4H),6.25(s,1H),4.90(s,1H),3.72(s,3H),3.58(ABq,2 H,J=14.3Hz),2.89−2.86(m,2H),2.78−2.76(m,2H),2.52−2.47 (m,2H),1.74(q,2H,J=5.2Hz),1.00(t,3H,J=5.2Hz).13C NMR(75MHz,CDCl3):δ171.2,170.4,149.7,134.7,133.8,130.0, 129.8,129.4,129.2,127.1,125.7,111.8,67.9,52.1,50.3,48.2,35.8, 25.0,18.2,13.5.ESI-MS m/z408.1[M+H]+.HRMS calcd for C20H23NO4SCl[M+H]+m/z408.1035,found408.1036.(S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl Pivalate(9d).To a solution of4a(337.5mg,1.0mmol,97.5%ee)in THF(15mL)was added Et3N(836μL,5.9mmol).After being stirred for10min,the mixture was cooled to0°C and pivaloyl chloride(738μL,6.1mmol)was added.After being stirred at25°C for4h,the mixture was poured into saturated sodium bicarbonate solution(60mL)and extracted with EtOAc(30mL).The organic layer was washed with brine,dried over anhydrous sodium sulfate,and concentrated under vacuum.The residue was purified by column chromatography to give the title compound in85%yield,99.1%ee(Chiral HPLC analytical conditions:same as those for9a).[α]D20+38.00°(c0.50,MeOH).1H NMR(300 MHz,CDCl3):δ7.69−7.23(m,4H),6.26(s,1H),4.90(s,1H),3.72 (s,3H),3.59(ABq,2H,J=14.1Hz),2.88−2.87(m,2H),2.79−2.77 (m,2H),1.30(s,9H).13C NMR(75MHz,CDCl3):δ175.1,171.2, 150.0,134.6,133.7,129.9,129.7,129.3,129.0,127.1,125.5,111.3, 67.7,52.0,50.2,48.1,39.0,26.9,24.9.ESI-MS m/z422.2[M+H]+. HRMS calcd for C21H25NO4SCl[M+H]+m/z422.1198,found 422.1193.(S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl2,2-Dimethylbutanoate (9e).Following a procedure similar to that described for the preparation of9d except that an equivalent amount of2,2-dimethylbutanoyl chloride was used in place of pivaloyl chloride,the title compound was obtained as a white solid in74.9%yield,mp98−100°C,99.5%ee(Chiral HPLC analytical conditions:same as those for9a).[α]D20+36.00°(c0.50,MeOH).1H NMR(300MHz,CDCl3):δ7.69−7.22(m,4H),6.25(s,1H),4.90(s,1H),3.71(s,3H),3.59 (ABq,2H,J=14.3Hz),2.88−2.87(m,2H),2.78−2.76(m,2H), 1.64(q,2H,J=7.3Hz),1.26(s,6H),0.88(t,3H,J=7.4Hz).13C NMR(75MHz,CDCl3):δ174.7,171.2,150.0,134.7,133.7,129.9, 129.7,129.4,129.1,127.1,125.5,111.4,67.8,52.1,50.3,48.1,43.1, 33.3,24.9,24.4,9.2.ESI-MS m/z436.2[M+H]+.HRMS calcd for C22H27NO4SCl[M+H]+,m/z436.1352,found436.1349.(S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl Benzoate(9f).Following a procedure similar to that described for the preparation of9a except that an equivalent amount of benzoic anhydride was used in place of acetic anhydride,the title compound was obtained as a white solid in 52.0%yield,mp84−86°C,93.5%ee(Chiral HPLC analytical conditions:same as those for9b).[α]D20+34.00°(c0.50,MeOH).1H NMR(300MHz,CDCl3):δ8.17−7.26(m,9H),6.42(s,1H),4.95 (s,1H),3.73(s,3H),3.68−3.57(m,2H),2.93−2.82(m,4H).13C NMR(75MHz,CDCl3)δ163.5,149.9,134.7,133.9,130.2,130.0, 129.8,129.5,128.6,128.5,127.2,125.9,112.1,67.8,52.2,50.4,48.2, 25.0.ESI-MS m/z442.1[M+H]+.HRMS calcd for C23H21NO4SCl [M+H]+m/z442.0891,found442.0880.(S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl4-Nitrobenzoate(9g).Fol-lowing a procedure similar to that described for the preparation of9d except that an equivalent amount of4-nitrobenzoyl chloride was used in place of pivaloyl chloride,the title compound was obtained as a yellow solid in26.0%yield,mp100−102°C,100%ee(Chiral HPLC analytical conditions:Chiralpak IC,4.6mm×250mm,eluting with 50%n-hexane+50%i-PrOH+0.1%Et2NH,flow rate0.5mL/min, oven temperature25°C,detection UV254nm).[α]D20+30.00°(c 0.50,MeOH).1H NMR(300MHz,CDCl3):δ8.18(s,4H),7.70−7.26(m,4H),6.47(s,1H),4.95(s,1H),3.73(s,3H),3.70−3.57 (m,2H),2.95−2.91(m,2H),2.84−2.82(m,2H).13C NMR(75 MHz,CDCl3):δ171.3,161.7,151.0,149.2,134.8,133.9,133.6,131.3, 130.0,129.8,129.5,127.2,126.6,123.8,112.6,67.8,67.3,52.2,51.6, 51.1,50.3,49.7,48.1,29.7,25.1.ESI-MS m/z487.0[M+H]+.HRMS calcd for C23H20N2O6SCl[M+H]+m/z487.0736,found487.0731. (S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl4-Methoxybenzoate(9h). Following a procedure similar to that described for the preparation of 9d except that an equivalent amount of4-methoxybenzoyl chloride was used in place of pivaloyl chloride,the title compound was obtained as a yellow oil in30.0%yield,96.9%ee(Chiral HPLC analytical conditions:same as those for9g).[α]D20+26.00°(c0.50,MeOH).1H NMR(300MHz,CDCl3):δ8.09−8.06(m,2H),7.68−7.26(m,4H),6.95−6.92(m,2H),6.38(s,1H),4.92(s,1H),3.85(s,5H),3.71−3.59(m,5H),2.90−2.79(m,4H).13C NMR(75MHz, CDCl3):δ171.2,164.5,163.1,149.9,134.6,133.7,132.7,132.3,129.9, 129.7,129.4,127.1,125.7,121.2,120.6,114.1,113.6,111.8,67.8,55.4, 52.0,50.3,48.1,25.0.ESI-MS m/z472.2[M+H]+,494.2[M+Na]+. HRMS calcd for C24H23NO5SCl[M+H]+m/z472.0993,found 472.0985.(S)-5-(1-(2-Chlorophenyl)-2-methoxy-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl2-Acetoxybenzoate(9i). Following a procedure similar to that described for the preparation。