高中化学奥赛有机第三讲烯烃
- 格式:doc
- 大小:337.00 KB
- 文档页数:8


高中化学奥林匹克竞赛辅导炔烃和二烯烃炔烃和二烯烃都是通式为C n H2n-2的不饱和烃,炔烃是分子中含有C≡C叁键的不饱和烃,二烯烃是含有两个C=C双键的不饱和烃,它们是同分异构体,但结构不同,性质各异。
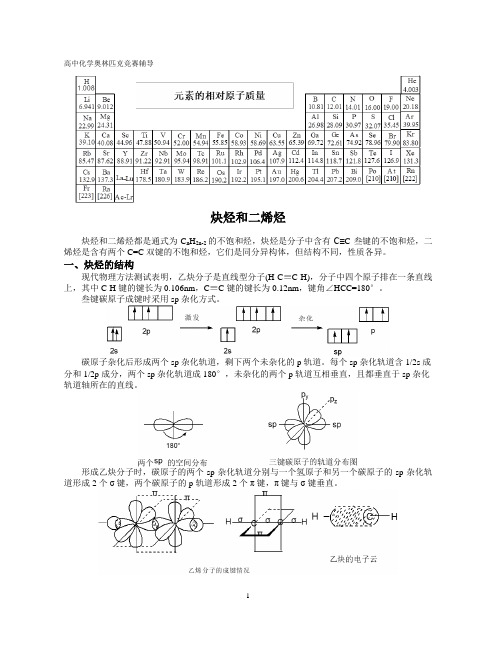
一、炔烃的结构现代物理方法测试表明,乙炔分子是直线型分子(H-C≡C-H),分子中四个原子排在一条直线上,其中C-H键的键长为0.106nm,C≡C键的键长为0.12nm,键角∠HCC=180°。
叁键碳原子成键时采用sp杂化方式。
碳原子杂化后形成两个sp杂化轨道,剩下两个未杂化的p轨道。
每个sp杂化轨道含1/2s成分和1/2p成分,两个sp杂化轨道成180°,未杂化的两个p轨道互相垂直,且都垂直于sp杂化轨道轴所在的直线。
形成乙炔分子时,碳原子的两个sp杂化轨道分别与一个氢原子和另一个碳原子的sp杂化轨道形成2个σ键,两个碳原子的p轨道形成2个π键,π键与σ键垂直。
二、炔烃的命名1.炔烃的系统命名法和烯烃相似,只是将“烯”字改为“炔”字。
2.烯炔(同时含有C=C和C≡C的分子)的命名:(1)选主链:选择含有C=C和C≡C的最长碳链作为主链。
(2)编号:从离C=C或C≡C最近的主链碳开始编号,并使C=C、C≡C位次之和最小。
如果链端与C=C、C≡C等距,则编号时通常使C=C双键具有最小的位次。
(3)命名:先命名烯烃,再命名炔烃。
如命名为(5R,2Z)-5-甲基-2-辛烯-6-炔。
三、炔烃的物理性质(1)随着碳原子数的增加,熔沸点逐渐升高,相对密度逐渐增大;碳原子数相同的炔烃,支链越多,熔沸点越低。
(2)碳原子数n≤4的炔烃为气态,其他的炔烃为液态或者固态。
(3)炔烃的相对密度一般比水的密度小。
(4)炔烃不溶于水,但易溶于有机溶剂。
四、炔烃的化学性质1.催化加氢:RC≡CR,+H2→RCH=CHR,,RC≡CR,+2H2→RCH2CH2R,。
催化加氢常用的催化剂为Pt、Pd、Ni,但产物一般难控制在烯烃阶段。

有机物烯烃,炔烃,醇,酚,糖等鉴别方法1. 烯烃、炔烃、二烯:能使溴的四氯化碳溶液,红色褪去,又能使高锰酸钾溶液,紫色褪去。
2.含有炔氢的炔烃: (1)能使硝酸银,生成炔化银白色沉淀(2)又能使氯化亚铜的氨溶液,生成炔化亚铜红色沉淀。
3.卤代烃:硝酸银的醇溶液,生成卤化银沉淀;不同结构的卤代烃生成沉淀的速度不同,叔卤代烃和烯丙式卤代烃最快,仲卤代烃次之,伯卤代烃需加热才出现沉淀。
4.小环烃:三、四元脂环烃可使溴的四氯化碳溶液褪色5.醇:(1)与金属钠反应放出氢气(鉴别6个碳原子以下的醇);(2)用卢卡斯试剂鉴别伯、仲、叔醇,叔醇立刻变浑浊,仲醇放置后变浑浊,伯醇放置后也无变化。
6.酚或烯醇类化合物:(1)用三氯化铁溶液产生颜色(苯酚产生兰紫色)。
(2)苯酚与溴水生成三溴苯酚白色沉淀。
7.羰基化合物:(1)鉴别所有的醛酮:2,4-二硝基苯肼,产生黄色或橙红色沉淀;(2)区别醛与酮用托伦试剂,醛能生成银镜,而酮不能;(3)区别芳香醛与脂肪醛或酮与脂肪醛,用斐林试剂,脂肪醛生成砖红色沉淀,而酮和芳香醛不能;(4)鉴别甲基酮和具有结构的醇,用碘的氢氧化钠溶液,生成黄色的碘仿沉淀。
8.甲酸:用托伦试剂,甲酸能生成银镜,而其他酸不能。
9.胺:区别伯、仲、叔胺有两种方法(1)用苯磺酰氯或对甲苯磺酰氯,在NaOH溶液中反应,伯胺生成的产物溶于NaOH;仲胺生成的产物不溶于NaOH溶液;叔胺不发生反应。
(2)用NaNO2+HCl:脂肪胺:伯胺放出氮气,仲胺生成黄色油状物,叔胺不反应。
芳香胺:伯胺生成重氮盐,仲胺生成黄色油状物,叔胺生成绿色固体。
10.糖:(1)单糖都能与托伦试剂和斐林试剂作用,产生银镜或砖红色沉淀;(2)葡萄糖与果糖:用溴水可区别葡萄糖与果糖,葡萄糖能使溴水褪色,而果糖不能。
(3)麦芽糖与蔗糖:用托伦试剂或斐林试剂,麦芽糖可生成银镜或砖红色沉淀,而蔗糖不能。
化学,。

高中化学有机物烷烃烯烃炔烃苯及苯的同系物卤代烃醇酚醛羧酸的化学性质一、烷烃的化学性质烷烃的化学性质很稳定,不与强酸、强碱、强氧化剂和强还原剂反应,在特定条件(有机化学的学习要特别注意反应条件)下能发生以下反应:1、取代反应2、氧化反应3、裂化和裂解大分子烷烃通过高温分解为小分子物质,如小分子烷烃、烯烃以及氢气。
二、烯烃的化学性质碳碳双键C=C是烯烃的官能团,烯烃化学性质比较活泼,容易发生加成、氧化还原,聚合:1、加成反应(1)1,2-加成A、丙烯和溴单质加成B、丙烯和溴化氢加成(马氏规则:H越多,越加H)(2)1,4-加成【注意】:发生1,2-加成或1,4-加成,取决于反应条件,一般低温倾向于发生1,2-加成,高温倾向于发生1,4-加成。
由此可见,相同的反应物在不同的条件下会生成不同的产物,因此要特别注意反应的条件,记准,记对!(3)环加成2、氧化反应(1)燃烧反应(2)高锰酸钾氧化书写步骤:A、碳碳双键断开变碳氧双键;B、双键碳上的氢原子变羟基。
(3)臭氧氧化只进行高锰酸钾氧化的第一步,C=C双键断裂变碳氧双键。
3、聚合反应4、烯烃的顺反异构两个双键碳原子上都连接两个不同的原子或原子团,就会有顺反异构。
顺式结构:两个相同原子或原子团在双键同一侧。
反式结构:两个相同原子或原子团在双键两侧。
三、炔烃的化学性质炔烃的官能团是碳碳三键,具有活泼的化学性质:1、加成反应炔烃可以和溴的四氯化碳溶液、卤素单质、氢气、氯化氢、水等发生加成反应。
2、氧化反应(1)燃烧:(2)能和高锰酸钾和臭氧反应(方程式不需要掌握)3、聚合反应聚乙炔中掺入某些物质,可以使其导电性显著增强,聚乙炔又叫做导电塑料。
四、苯及其同系物的化学性质1、取代反应(2)苯不能使酸性高锰酸钾溶液褪色,也不能与溴水发生加成反应使溴水褪色,但苯能将溴从溴水中萃取出来。
(3)苯的同系物的氧化反应五、卤代烃的化学性质1、NaOH的水溶液发生取代反应变成醇2、NaOH的醇溶液发生消去反应变成烯(札依采夫规则)【注意】:札依采夫规则:H越少,越减H。

专题三烯烃§1.烯烃的结构、异构及命名一、烯烃的结构乙烯是最简单的烯烃,分子式为C 2H 4,构造式H 2C=CH 2,含有一个双键C=C,是由一个σ键和一个π键构成。
杂化轨道理论设想碳原子成键时,由一个s 轨道和两个p 轨道进行杂化,组成三个等同的sp 2杂化轨道,sp 2轨道对称轴在同一平面上,彼此成1200角.此外,还剩下一个2p 轨道,它的对称轴垂直于sp 2轨道所在的平面.C-C σ键在乙烯分子中,两个碳原子各以一个sp 2轨道重叠形成一个C-C σ键,又各以两个sp 2轨道和四个氢原子的1s 轨道重叠,形成四个C-H σ键,五个σ键都在同一平面上。
每个碳原子剩下的一个p 轨道,它们平行地侧面重叠,便组成新的分子轨道,称为π轨道。
1.π键的特点:⑴π键重叠程度比σ键小,不如σ键稳定,比较容易破裂。
碳碳π键的键能等于264.4kJ/mol 。
[610(C=C 键能)-345.6(C-C 键能)]小于C-C 单键的键能345.6kJ/mol.⑵π键具有较大的流动性,容易受外界电场的影响,电子云比较容易极化,容易给出电子,发生反应。
这是由于π键的电子云不象σ键电子云那样集中在两原子核连线上,而是分散成上下两方,故原子核对π电子的束缚力就较小。
2.C=C 和C-C 的区别:⑴C=C 的键长比C-C 键短。
两个碳原子之间增加了一个π键,也就增加了原子核对电子的吸引力,使碳原子间靠得很近。
C=C 键长0.134nm,C-C 键长0.154nm 。
⑵C=C 两原子之间不能自由旋转。
由于旋转时,两个p 轨道不能重叠,π键便被破坏。
§2.烯烃的制备1.卤代烷脱卤化氢由一卤代烷制备烯烃,要用强碱作试剂。
伯卤代烷永叔丁基醇钾在二甲亚砜(DMSO ,即二甲硫氧)溶液中进行反应,效果最好。
CH 3(CH 2)15CH 2CH 2Cl (CH 3)3COK DMSOH 3C(H 2C)15HC CH 21-chlorooctadecaneoctadec-1-ene仲、叔卤代烷形成烯烃时,其双键位置主要趋向于在含氢较少的相邻碳原子上。
例如:H 3CH 2CC CH 3BrCH 3KOH/EtOH H 3CHC C(CH 3)2+C CH 2H 3CH 2CCH 371%29%以生成取代较多的烯烃为主要产物,这就是札依切夫(Saytzeff)规律。
(1875年由A.M.扎伊采夫提出。
在醇脱水或卤代烷脱卤化氢中,如分子中含有不同的β—H 时,则在生成的产物中双键主要位于烷基取代基较多的位置,即含H 较少β碳提供氢原子,生成取代较多的稳定烯烃。
) 2.醇脱水醇在无机酸催化剂存在下加热时,失去一分子水而得到相应的烯烃。
常用的酸是硫酸和磷酸。
CH 3CH 2OHH 2SO 4,170O CH 2C CH 2etheneethanolOHH 2SO 4,140O C(Z)cyclohexanolcyclohexeneH 3CC CH 3OH CH 320%H SO ,85OC(H 3C)2C CH 22-methylpropan-2-ol 2-methylprop-1-ene3.脱卤素C XC X+ ZnCC+ ZnX 2H 3CH C Br HC BrCH 3+ Zn H 3CHC CHCH 3but-2-ene2,3-dibromobutane这个反应可用来保护双键。
当要使烯烃的某一部位发生反应时,可先将双键加卤素,随后用锌或镁处理使双键再生。
§3.消除反应卤代烃的脱卤化氢和醇脱水反应都是消除反应。
可分为:1.2-消除反应:1,1-消除反应1,3-消除反应 一、β-消除反应1.消除反应的历程-E2、E1和E1cb(1)单分子消除反应(E1)υ=k[RX]反应速率只与反应物浓度有关,而与碱性试剂浓度无关。
特点:E1历程是一个一级反应,由于它通过正碳离子所以会有重排现象产生。
碳正离子能重排成更稳定的碳正离子。
⑵、双分子消除(E2)bimolecularelimination由碱性EtO-进攻β-H,形成过渡态,β-H被夺去。
分子中的离去基团Br带着一对电子离去。
α与β碳原子形成双键,协同进行。
υ=k[CH3CH2CH2Br][CH3CH2O-]二级反应特点:二级反应,没有重排现象。
E2有显着的同位素效应。
动力学同位素效应:所谓动力学同位素效应,即反应物分子中的某元素被其同位素取代后反应速度发生变化。
最常用的体系是重氢(D)取代氢(H),这种同位素效应用K H/K D表示。
使C-H键破裂所需的活化能要比使C-D键破裂所需的活化能小,因此,在反应中C-H键破裂的速度比C-D键快。
这种动态同位素效应可以用两种反应的速度常数来衡量。
动态同位素效应=k H/k D说明了在决定速率步骤的过渡态中有C-H或C-D键的断裂。
一级动力学同位素效应粗略计算在室温(298K)k H/k D=6.5升高反应温度同位素效应(k H/k D)将会降低。
由于H比D轻一倍,因而断裂C-H键比断裂C-D键快得多。
E1动态同位素效应为1左右。
(3)、共轭碱历程(Conjugatebase)Elcb在碱进攻下,H首先离去,然后L再以负离子形式离去。
因为碳负离子中间体是反应物的共轭碱故称共轭碱历程。
当β-碳原子上连有吸电子基团时有利于E1cb历程进行。
因为吸电子基有利于质子转移给碱,又使形成的碳负离子稳定。
另外这个吸电子基团应不具有离去集团的性质。
同时C-L键必须不容易离解。
简单的卤代烷和磺酸酯中现测不到这种机理。
E1cb历程不常见。
只有当离去基邻位有等能稳定碳负离子的官能团时,E1cb才能发生。
2.消除反应的取向在脱卤化氢及醇脱水反应中,所生成的烯烃都遵守扎依采夫规则,即优先形成具有较多烷基取代的烯烃。
⑴E2:如:现在一般用过渡态理论来解释:由于过渡态已具有部分烯烃的性质,因此在产物中可使烯烃稳定的因素也应使其相应的过渡态稳定。
Ⅰ比Ⅱ有较大的超共轭效应,因此Ⅰ较稳定,形成时所需的活化能小,所以,消除主要产物是2-戊烯⑵E1:E1反应中决定取向的是在第二步,可以预料在碳正离子形成过程中,不论离去基团是什么,取向基本上是一样的,占优势的产物是取代基较多的烯烃。
如:⑶几个特殊例子:由于C-F键较牢,不易断裂,因而在它的过渡态中只有少许烯烃的特征而有大量的负碳离子的特征。
所以,在这里伯氢将优先为碱所夺取,因为(a).伯碳位阻小,容易被碱进攻;(b).碳负离子的稳定性和正碳离子时相反,即10>20>30,所以氟代物的消除取向和其他的卤代物相反。
碱的体积大小对取向也有影响,(CH3)3C-O-比CH3O-体积大,所以它夺取末端的伯氢较夺取中间的仲氢为易(空间位阻小)。
3.消除反应的立体化学许多实验事实证明,大部分E2反应是反式消除。
反应时分子中是H和L彼此处于反式共平面。
如:这是因为,根据E2历程,d2>d1,碱与离去基团排斥力较小。
这样有利用B:接近H将它拉下来根据形成π键的要求,最好过渡态中氢和离去基团是在相同的平面中。
这样能使部分形成的双键中的p轨道有较多的重迭。
为了达到同平面的要求,H与X处于反式的构象比处于顺式的构象容易形成一些。
因为两个相邻基团间的vanderwaals斥力较小,所以一般消除反应都是反式消除形式进行。
如:例:化合物(1)、(3)在发生消除反应时,为什么(1)比(3)反应速度慢的多?化合物(1)最稳定的构象为(5),其中三个体积大的取代基-Cl、-CH3、-CH(CH3)2都以e键与环相连,起消去反应时要变成(1)才能使C-Cl为a键,这时仲碳原子上的C-H在a键的位置。
因此,产物(2)即Hofmann烯烃在(3)的最稳定的构象中C-Cl已为a键,它的两边都有a氢原子,因此,可生成两种烯烃。
因此例符合Saytzeff 规律。
化合物(1)起反应要先从稳定的构象变成不稳定的构象(1),这需要能量,因此,反应速度比(3)慢,。
因此,(1)比(3)的反应速度慢的多。
这里我们要指出:虽然大部分E2反应是反式消除的,可是也会有顺式消除,特别是在一些环状体系中。
如:关于E1反应的立体化学,没有一定的规律。
顺式、反式消除都有,两者的比例随作用物而不同。
4.消除反应与亲核取代反应的竞争消除反应与亲核取代反应都是由同一亲核试剂的进攻引起的。
进攻α碳就是取代,进攻β-氢原子就引起消除,这两种反应常常是同时发生和互相竞争的。
那么我们能否控制这两个并存而且相互竞争的反应呢?看来完全控制还不能,但是已有一些规律的东西可供我们工作时参考。
⑴反应物的结构制烯时最好用30卤代物,制醇时最好用10卤代物。
因为卤代物的结构对消除和取代反应有如下的影响。
⑵试剂的碱性碱性强,浓度大,越有利于消除反应,碱性弱,亲核强,浓度较小,则有利于取代反应。
如:NH3有亲核能力,但碱性不大。
因而,它只能进行亲核取代反应,而不能进行消除反应,若要发生消除反应则采用强碱-NH2。
⑶溶剂的极性一般来说,增加溶剂的极性有利于取代反应,不利于消除反应。
所以常用KOH的水溶液从卤代烷制醇,而用它的醇溶液制烯烃。
这是由于取代反应中过渡态的负电荷分散比消除反应中过渡态的负电荷分散为小,所以高极性溶剂对取代反应中过渡态的稳定作用比较大些,因较有利于取代反应。
(电荷集中溶剂化作用好,电荷分散溶剂化作用差,溶剂化强的就稳定)。
醇溶液(极性较小)-有利于制备烯烃,水溶液(极性大)-有利于制备醇。
⑷反应温度升高温度将提高消除反应的比例。
这是由于在消除过程中涉及C-H键的断裂,活化能较高,升高温度对它有利。
如虽然提高温度使取代反应加快,可是对它的影响没有消除反应那么大,所以提高反应温度将增加消除产物。
二、α-消除反应1.卡宾(carbenes)的产生如:2.卡宾的结构状态数=2|S|+1,S:自旋量子数之和单线态较不稳定,常常是在分解初期首先产生的形式,在液相中最初形成的卡宾。
在气相中,尤其在惰性气体(N2或Ar)存在下单线态经碰撞而失去能量后转变为三线态,三线态能量比单线态低42KJ/mol。
3.卡宾的反应⑴与C=C双键的加成:在液态顺-2-丁烯中,卡宾的加成只生成顺-1,2-二甲基环丙烷,对反-2-丁烯加成只生成反-1,2-二甲基环丙烷,这种立体专一的顺式加成,一般认为是单线态卡宾对双键π电子对的协同反应。
单线态的空轨道是缺电子的,因此是亲电的,他能从烯烃π键获得电子,立体化学结果表明它是同时连接到双键的两个碳原子上去的。
在气态,卡宾与2-丁烯(顺式或反式)的加成产物是等量的顺和反-1,2-二甲基环丙烷的混合物。
参加反应的卡宾是三线态,非立体选择性。
从上述反应可以看出,单线态卡宾和烯烃的加成是协同反应(顺式加成),而三线态卡宾和烯烃的加成都是按分步的双游离基理发生反应。