动植物重测序
- 格式:pdf
- 大小:9.21 MB
- 文档页数:23

动植物基因组de novo常见问题基础知识1、什么是基因组de novo测序答:对某一物种进行高通量测序,利用高性能计算平台和生物信息学方法,在不依赖于参考基因组的情况下进行组装,从而绘制该物种的全基因组序列图谱。
2、普通基因组的定义答:单倍体,纯合二倍体或者杂合度<%,且重复序列含量<50%,GC 含量为35%到65%之间的二倍体。
3、复杂基因组的定义答:杂合率>%,重复序列含量>50%,GC含量处于异常的范围(GC 含量<35%或者GC含量>65%=的二倍体,多倍体。
诺禾致源对二倍体复杂基因组进一步细分为微杂合基因组(%<杂合率<%=、高杂合基因组(杂合率>%)以及高重复基因组(重复序列比例>50%)。
4、怎么查询基因组的大小答:查询植物基因组大小的网站:;查询动物基因组大小的网站:。
、5、基因组的项目周期6、基因组承诺的组装指标答:简单基因组:contig N50>20K,scaffold N50>500K;复杂基因组:contig N50>20K,scaffold N50>300K。
样品要求1、动植物基因组测序对取样有什么要求答:植物:需要黑暗无菌条件下培养的黄化苗、组培苗,基因组样本量500μg~1mg,越多越好。
选择纯合或杂合度尽可能小的样品(杂合度<%)。
动物:应选取肌肉、血液等含脂肪较少的部位取样,尽量选择同一个体取样,以减少个体差异性对后续拼接的影响。
基因组样本量500μg~1mg,越多越好。
样本的性别决定模式是XY型,则尽量选择雌性个体(XX型),如果是ZW型,则尽量选择雄性个体(ZZ型)。
2、全基因组测序对DNA样本有什么要求答:(1)样品需求量(单次):小片段文库,≥3μg;2Kb~5Kb大片段文库,≥20μg;10Kb~20Kb大片段文库,≥60μg;完成全基因组测序样品DNA量需求约为500μg~1mg;(2)样品浓度:对于小片段文库,≥50ng/μl,对于2Kb~5Kb 大片段文库,≥150ng/μl;对于10Kb~20Kb大片段文库,≥150ng/μl;(3)样品纯度:OD260/280=~;无蛋白质、RNA污染或肉眼可见杂质污染;(4)样品质量:基因组完整。

高通量测序技术在动植物研究领域中的应用一、本文概述随着生物技术的飞速发展,高通量测序技术(High-throughput sequencing technology)已成为动植物研究领域的重要工具。
该技术以其快速、准确、高效的特点,极大地推动了动植物基因组学、转录组学、表观遗传学等多个研究领域的进步。
本文旨在全面综述高通量测序技术在动植物研究领域的应用,包括动植物基因组测序、基因表达分析、基因功能研究、种质资源鉴定、遗传育种以及生态保护等方面。
通过深入剖析这些应用案例,旨在为读者提供一个清晰、全面的高通量测序技术应用全景,以期推动该技术在动植物研究领域的进一步发展和应用。
二、高通量测序技术的基本原理与方法高通量测序技术,又称为下一代测序技术(Next Generation Sequencing,NGS),是近年来生物信息学领域的一项革命性技术。
其基本原理是通过将待测样本的DNA或RNA片段化,然后利用高通量测序平台对这些片段进行大规模并行测序。
这种方法大大提高了测序速度和效率,降低了成本,使得研究者可以对基因组、转录组甚至单细胞进行全面的深入研究。
高通量测序的方法主要包括样本准备、文库构建、测序及数据分析等步骤。
在样本准备阶段,研究者需要从动植物组织中提取高质量的DNA或RNA,并通过特定的酶处理将其片段化。
文库构建则是将这些片段与测序引物连接,形成适合测序的文库。
测序阶段则通过高通量测序仪器对文库进行大规模的并行测序,得到原始的测序数据。
在数据分析阶段,研究者需要使用生物信息学工具对原始数据进行处理、组装和注释,最终得到基因组的序列信息、基因结构、表达水平等关键信息。
通过这些信息,研究者可以对动植物的基因组结构、功能、进化等方面进行深入的研究。
高通量测序技术在动植物研究领域的应用广泛,包括但不限于基因组测序、转录组测序、表观遗传学研究、单细胞测序等。
这些应用不仅有助于我们更深入地理解动植物的生物学特性,也为动植物育种、疾病防治、生态保护等领域提供了新的思路和方法。
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS (Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
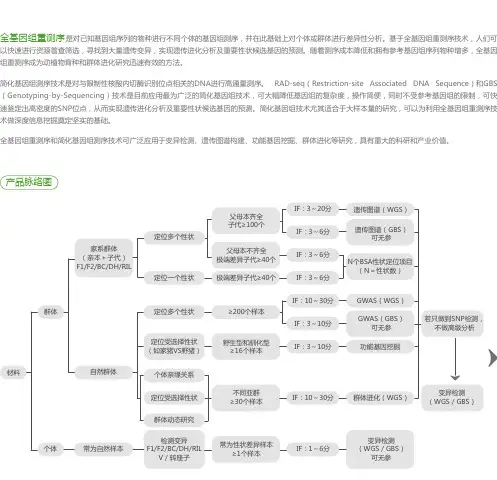
产品脉络图。


动植物多样性调查方法总结摘要:动植物多样性调查是保护和管理生物资源的重要工作。
本篇文章将总结动植物多样性调查的常用方法和技术,包括样方法、线路法、点数法、红外相机技术和DNA条形码技术等。
这些方法可以提供科学依据,促进生物多样性保护和环境监测。
引言:生物多样性是地球上生命的重要组成部分,对维持生态平衡和人类福祉有着重要影响。
为了保护和管理生物多样性,了解物种分布和数量就显得尤为重要。
动植物多样性调查是研究和监测生物多样性的重要手段之一。
本文将介绍动植物多样性调查的一些常用方法和技术,希望能够对相关研究和保护工作起到一定的指导作用。
一、样方法样方法是动植物多样性调查中最常用的方法之一。
该方法通过在特定的地理区域内设置样方,统计不同物种的数量和分布情况。
样方的选择要考虑样方的大小、形状和数量等因素,以确保调查结果的代表性。
一般情况下,研究区域会被划分成多个子样方,每个样方内的调查时间和频率也要一致,以减小误差。
二、线路法线路法是用于调查动植物分布和数量的重要手段。
这种方法通过在待调查地区的特定路线上进行调查,记录沿途发现的物种和数量。
与样方法相比,线路法具有较高的空间尺度和连续性,可以揭示物种的扩散和迁移规律。
同时,线路法的样本量相对较大,可以减小随机误差,提高数据的可靠性。
三、点数法点数法是一种快速调查动植物多样性的方法。
该方法通过在待调查区域内随机或系统地设置调查点,记录每个调查点上出现的物种和数量。
点数法相对于样方法和线路法来说,调查时间和精力要求较少。
但是,点数法的空间尺度相对较小,可能无法全面反映调查区域的多样性。
因此,在应用点数法时,需要合理设置调查点的数量和分布。
四、红外相机技术红外相机技术是近年来研究动植物多样性调查中的创新方法之一。
这种技术通过设置红外相机在待调查地区进行连续监测,捕捉到的照片可以反映物种的活动和数量。
红外相机技术可以避免人为因素对调查结果的干扰,同时也可以在夜间或难以进入的地区进行调查。

植物重测序项目完成的主要指标通常包括但不限于以下几个方面:
1.测序深度(Coverage):达到预设的测序深度是重测序项目成功的关键指标之一。
例如,一般要求覆盖整个基因组的平均深度至少为10X或更高,对于复杂区域或重点研究区域,可能需要达到更高的深度,如30X或50X。
2.数据质量(Data Quality):数据质量包括测序reads 的质量评分(如Phred quality score)、reads 的长度、GC含量分布、错误率等。
高质量的数据意味着序列读段的准确度高,可以更准确地反映基因组的真实信息。
3.序列比对率(Mapping Rate):比对到参考基因组的序列占总序列的比例,通常期望达到90%以上。
高比对率意味着大部分测序数据得到了有效利用,可以用于后续的变异检测和分析。
4.变异检测准确性(Variant Calling Accuracy):发现和鉴定SNPs(单核苷酸多态性)、InDels(插入/缺失突变)、结构变异(Structural Variants, SVs)等遗传变异的准确性,通常通过与已知数据库或验证实验进行对比,评估变异检出的假阳性率和假阴性率。


全基因组重测序基础及高级分析知识汇总借着2月8号,刚刚在Nature上发表柑橘的遗传进化的文章,小编来讲述一下全基因组重测序基础知识,以及常见的分析思路及软件,帮助大家迅速入门。
全基因组重测序是通过对已有参考序列(Reference Sequence)的物种的不同个体进行基因组测序,并以此为基础进行个体或群体水平的遗传差异性分析。
通过全基因组重测序,研究者可以找到大量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插入缺失(InDel,Insertion/Deletion)、结构变异(Structure Variation,SV)等变异位点。
基于以上变异位点作为分子遗传标记,在人类复杂疾病、动植物经济性状和育种研究及物种起源、驯化、群体历史动态等方面具有重大的指导意义(Bentley2006; Casillas& Barbadilla 2017)。
一、基础理论知识全基因组重测序研究主要是依据在全基因组水平发现的分子遗传标记进行物种的群体遗传学研究,进一步的利用统计方法进行影响表型和经济性状候选基因和功能突变的研究。
分子群体遗传学研究的理论基础知识及统计分析方法日趋完善和呈现多样性,作为初学者,有必要对其中的一些基础概念有一定的了解,才能为后续的深入学习、研究提供基石。
以下基础知识主要参考国内动物遗传学书籍和最新的一篇关于分子群体遗传学方面的综述改变而成(吴仲贤编1961; 李宁2011; 吴常信2015; Casillas & Barbadilla 2017)。
高通量测序技术作为分子群体遗传学研究的有力工具,在科学研究、生产及疾病诊断治疗中起到原来越重要的作用,对关于高通量测序相关的理论基础知识进行一定程度的了解,也有助于文献阅读和。
1. 群体遗传学基础知识群体(Polulation):是指生活在一定空间范围内,能够相互交配并生育具有正常生殖能力后代的同种个体群。

基因测序技术在动植物保护中的应用近年来,随着科技的不断发展,基因测序技术也逐渐成为了一个热门话题。
基因测序技术是一项精确的科技,它可以有效地揭示生物的基因信息,进而为动植物保护提供有力的支撑。
在本文中,我们将探讨基因测序技术在动植物保护中的应用。
一、动物保护动物保护是基因测序技术在动物领域中的主要应用之一,它可以帮助我们更好地了解动物的基因信息,从而更好地保护野生动物的生命安全。
基因测序技术可以帮助我们更好地理解种群的演化,揭示动物的遗传信息,为保护濒危物种提供有效的保护策略。
例如,通过基因测序技术,科学家们可以研究黑猩猩、大猩猩等灵长类动物的基因序列,了解其遗传信息,从而更好地率领我们保护这些珍稀动物。
此外,基因测序技术还可以帮助我们分析海豚、鲸鱼等海洋生物的基因组信息,不仅可以了解它们的遗传特征,更有助于发现海洋生物的疾病,为疾病治疗带来新的思路和方法。
同时,基因测序技术还能够为野生动物保护提供更加精准的基因监测和管理方式,从而更好地掌握种群信息,为野生动物的保护工作做出贡献。
二、植物保护除了在动物领域中发挥积极的作用外,基因测序技术在植物保护领域中也有着广泛的应用。
植物测序可以帮助我们了解植物的遗传信息,从而更好地研究植物的适应性以及如何为植物提供更好的生长环境。
例如,基因测序可以帮助我们了解植物基因的序列,并发现植物中的一些新基因,加深我们对植物基因的认识。
此外,还可以发现一些新的植物物质,控制植物生长的遗传调控过程,为农业生产带来创新的思路和方法。
同时,基因测序技术还可以帮助我们研究植物抗性,了解植物的适应性和环境适应性,为植物保护工作提供基础数据和科学依据。
三、基因编辑技术基因编辑技术是一种新兴的遗传工程技术,它可以对生物基因进行精准编辑,以期改善生物的遗传性状。
基因编辑技术可以帮助我们解决动植物遗传性状的问题,对健康和生命的保护有着深远的意义。
例如,在动物保护领域中,基因编辑技术可以帮助我们增强野生动物的免疫力、减轻动物的痛苦或解决某些遗传性状的问题。

动植物Denovo测序知识⼤讲解⾼通量测序的技术开起我们探索动植物基因组奥秘的步伐,提到动植物基因组测序,这就不得不提⼀个概念——de novo测序。
那么什么是de nove测序呢,它与重测序有什么区别呢?De nove测序中Read、Contig和Scaffold等⼜代表什么呢?De nove测序中为什么要建不同⼤⼩⽚段的梯度⽂库?基因注释⼜是注释哪些内容?各位客官别急,且听⼩编给您细细讲来。
1De novo测序概念De novo是⼀个拉丁⽂,代表从头开始的意思,⽽de nove测序则是指在不需要任何参考序列的情况下对某⼀物种进⾏基因组测序,然后将测得的序列进⾏拼接、组装,从⽽绘制该物种的全基因组序列图谱。
由于⾼通量测序长度的限制,⽬前测序策略是先将基因组打断⼩的⽚段,然后再对测出序列⽚段进⾏拼接,最终得到物种的序列图谱如图1所⽰。
图1 ⾼通量测序模式图2De novo测序与重测序区别重测序概念:重测序是全基因组重新测序的简称,是指是对已知基因组序列的物种进⾏不同个体的基因组测序,并在此基础上对个体或群体进⾏差异性分析。
从概念上来看两者的区别在于de nove测序是对没有参考基因组的物种进⾏测序,⽽重测序是对已有基因组的物种进⾏测序,这只是它们区别很⼩的⼀部分。
从原理上来看de nove测序和重测序最根本的区别在于de nove测序需要对测序得到的Reads进⾏拼接组装,⽽重测序得到的数据则是没有组装的短的Reads序列。
值得注意的是,随着测序成本的降低以及组装算法的改进,de nove测序成本越来越低,⽬前来说de nove测序不只对于没有参考基因组物种进⾏测序,还可以对⼀些特有的亚种、品种以及变种等进⾏测序。
3Reads Conting Scaffold概念Reads:即我们通常说的读长的意思,它是指⾼通量测序平台直接产⽣的DNA序列。
Contig:是指Reads基于Overlap关系,拼接获得的长的序列;Scaffold:是指将获得的Contig根据⼤⽚段⽂库的Pair-end关系,将Contig进⼀步组装成更长的序列;关于三者之间的关系如图2所⽰,注意的是Contig是⽆Gap的连续的DNA序列,⽽Scaffold是存在Gap的DNA序列。

–基于PCR和DNA测序技术快速鉴定动植物品种随着科学技术的不断发展,生物学研究方法也在不断革新。
PCR(聚合酶链反应)和DNA测序技术成为了现代生物学领域中最为重要的实验方法之一。
这些技术不仅在基础研究中发挥着重要作用,而且被广泛应用于医疗、食品安全以及动植物品种鉴定等领域。
本文将着重介绍基于PCR和DNA测序技术快速鉴定动植物品种的原理和应用。
一、PCR技术在动植物品种鉴定中的应用PCR技术是一种体外扩增DNA片段的方法,它能够在短时间内产生大量目标DNA片段,从而方便后续的分析与鉴定。
在动植物品种鉴定中,PCR技术可以通过特异性引物选择性扩增目标基因片段,从而鉴定物种的遗传特征。
以下是PCR技术在动植物品种鉴定中的应用案例:1. 动物品种鉴定动物的基因组中有许多特定的DNA序列可以用于鉴定品种。
例如,鸟类常用的线粒体DNA序列和核DNA序列可以用来快速鉴定不同品种的鸟类。
犬种鉴定是另一个应用广泛的领域,通过PCR技术扩增犬的线粒体DNA或核DNA,可以快速准确地鉴定出犬的品种。
2. 植物品种鉴定植物基因组中的DNA序列对于鉴定物种也起着重要作用。
例如,树木的木质部DNA序列可以用于鉴定树木的物种。
此外,某些作物栽培品种在基因组中具有特定的性状基因,通过PCR技术鉴定这些基因的存在与否可以确定作物品种。
二、DNA测序技术在动植物品种鉴定中的应用DNA测序技术是指通过测定DNA分子中的碱基序列,从而获取DNA信息的方法。
这项技术的出现,使得我们能够更准确地确定DNA序列,从而实现更精确的物种鉴定。
以下是DNA测序技术在动植物品种鉴定中的应用案例:1. 动物品种鉴定DNA测序技术可以通过测定某些保守基因的碱基序列,来确定不同动物品种间的差异。
例如,通过测序线粒体DNA的D-loop区域,可以判断不同物种的遗传差异,从而进行物种鉴定和进化研究。
2. 植物品种鉴定植物基因组中的DNA序列也可以通过测序来实现品种鉴定。
2013.072013.102014.092014.112015.04图1 异源多倍体棉花基因组共线性分析与非对称进化分析图2 MYB基因家族表达模式分析, Jiang W, et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement [J]. Nature Biotechnology, 2015, 33(5): 531-537.图3 金丝猴植食性机制的分析图4 金丝猴有效群体大小分析参考文献Zhou X, Wang B, Pan Q, Li R, Li M. Whole-genome sequencing of the nub-nosed monkey provides insights into folivory and evolutionary history [J]. Nature Genetics, 2014, 46(12):1303-1310.图5 藏猪及其它猪种的群体遗传结构分析参考文献Li M, Tian S, Jin L, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars [J]. Nature genetics, 2013, 45(12): 1431-1438.图6 进化分析结果图7 脂肪酸能量代谢途径蓝色表示正选择基因;红色表示特异性基因参考文献Qu Y, Zhao H, Han N, et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau [J]. Nature communications, 2013, 4.图1 7株野生大豆共有和特有基因集图2 野生大豆开花时间调控基因SNP和InDel变异参考文献Li Y, Zhou G, Ma J, Jiang W, Li R#, et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits [J]. Nature biotechnology, 2014.32(10):1045-1052.图3 部分novel sequence在世界人群中的分布参考文献Li R, Li Y, Zheng H, et al. Building the sequence map of the human pan-genome [J]. Nature biotechnology, 2010, 28(1): 57-63.。
重测序参考手册目录目录 (1)1. 重测序简介 (3)2. 重测序实验方法 (3)基因组DNA抽提 (3)基因组DNA样品建库 (3)上机前定量 (4)3. 重测序分析内容 (4)重测序分析流程 (5)重测序分析内容 (5)4. 重测序重要技术参数 (6)5. 重测序分析内容解释 (6)6. 重测序分析内容示例 (6)SNP、INDEL的样本差异分析 (12)7. 成功分析案例/或已发表论文 (14)8. 概念及常用工具链接 (14)1. 重测序简介全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
全基因组重测序的个体,通过序列比对,可以找到大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion/Deletion)、结构变异位点(SV,Structure Variation)位点。
众信可以协助客户,通过生物信息手段,分析不同个体基因组间的结构差异,同时完成注释。
2. 重测序实验方法提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段(0.2~5Kb),加上接头, 进行cluster制备(Solexa)或E-PCR (SOLiD),最后利用Paired-End或者Mate-Pair的方法对插入片段进行重测序。
实验步骤主要包括以下几点:基因组DNA抽提不同生物(植物、动物、微生物)的基因组DNA的提取方法有所不同; 不同种类或同一种类的不同组织因其细胞结构及所含的成分不同,分离方法也有差异。
在提取某种特殊组织的DNA时必须参照文献和经验建立相应的提取方法, 以获得可用的DNA大分子。
尤其是组织中的多糖和酶类物质对随后的酶切、PCR反应等有较强的抑制作用,因此用富含这类物质的材料提取基因组DNA时, 应考虑除去多糖和酚类物质。
基因组DNA样品建库这是样品准备过程中最主要的环节,也就是真正意义上的建库(通常我们所说的建库包括整个样品准备的过程)。
首页 科技服务 测序指南 基因课堂 市场活动与进展 文章成果 关于我们全基因组选择1. Meuwissen T H, Hayes B J, Goddard M E.Prediction of total genetic value using genome-wide dense marker maps[J]. Genetics, 2001, 157(4): 1819 1829. 阅读原文>>2. Haberland A M, Pimentel E C G, Ytournel F, et al. Interplay between heritability, genetic correlation and economic weighting in a selection index with and without genomic information[J]. Journal of Animal Breeding and Genetics, 2013, 130(6): 456-467. 阅读原文>>3. Wu X, Lund M S, Sun D, et al. Impact of relationships between test and training animals and among training animals on reliability of genomic prediction[J]. Journal of Animal Breeding and Genetics, 2015, 132(5): 366-375. 阅读原文>>4. Goddard M E ,Hayes BJ. Genomic selection [J]. Journal of Animal Breeding and Genetics,2007,124:323:330. 阅读原文>>5. Heffner E L, Sorrells M E, Jannink J L. Genomic selection for crop improvement [J]. Crop Science, 2009, 49(1): 1-12. 阅读原文>>参考文献全基因组选择简介Meuwissen等[1]在2001年首次提出了基因组选择理论(Genomic selection , GS),即利用具有表型和基因型的个体来预测只具有基因型不具有表型值动植物的基因组育种值(GEBV)。
重测序bsa技术原理
重测序bsa技术原理是一种基于高通量测序技术的分析方法,通过对DNA或RNA序列进行重测序,可精确鉴定样本中存在的基因变异、单核苷酸多态性等遗传变异信息,为个体化医疗提供重要参考。
重测序bsa技术主要分为两个步骤:首先,对样本进行高通量测序,生成原始测序数据;然后,通过数据分析筛选出与目标性状相关的遗传变异,进而确定与性状相关的基因位点。
在数据分析方面,重测序bsa技术采用了一系列生物信息学工具和统计分析方法,包括比对、变异检测、基因型调用、单核苷酸多态性分析等。
其中,比对是重要的一环,即将原始测序数据与参考基因组进行比对,以确定样本中存在的遗传变异信息。
与传统的遗传分析方法相比,重测序bsa技术优势明显。
首先,在检测精度上,重测序bsa技术能够检测到低频率的变异信息,具有更高的灵敏度;其次,在样本处理上,重测序bsa技术不需要对样本进行前期处理,可直接对样本进行测序,降低了操作的复杂性和成本;最后,在应用范围上,重测序bsa技术不仅可应用于动植物遗传分析、人类遗传疾病筛查等领域,还可广泛应用于农业生产、环境保护等领域。
总之,重测序bsa技术是一种高效、准确、广泛应用的遗传分析方法,将为基因组学研究和个体化医疗提供重要支持。
- 1 -。
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS(Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
产品脉络图动植物重测序建库测序单个性状家系群体自然群体SNP/InDel/SV/CNV/转座子基因组DNA有效SNP性状定位群体进化群体进化(基于简化基因组测序) 群体进化(基于全基因组重测序) 变异检测(基于简化基因组测序)SNP检测/SSR检测遗传图谱全基因组关联分析(GWAS)功能基因挖掘变异检测(基于全基因组重测序) QTL定位BSA性状定位多个性状动植物重测序动植物重测序概述SNP检测、注释及统计基因组DNA350 bp小片段文库HiSeq PE150测序数据质控与参考基因组比对利用全基因组重测序技术对某一物种个体或群体的基因组进行测序及差异分析,可获得SNP、InDel、SV、CNV、PAV、转座子等大量的遗传多态性信息,建立遗传多态性数据库,为后续揭示进化关系、功能基因挖掘等奠定基础。
基于全基因组重测序技术进行变异检测,可在全基因组范围内精确检测多种变异,与传统的分子标记和芯片相比,具有周期短、密度高、检测全面、性价比高等技术优势。
诺禾致源使用的检测软件及参数为国际高影响因子文章通用标准,对获得的变异进行全方位评估以确保变异准确、验证率高。
变异检测(基于全基因组重测序)技术路线技术参数DNA样品量: ≥3 μg测序策略:每个个体测序深度10-30X,推荐深度SNP、InDel(≥10X),SV(≥20X),CNV(≥30X)转座子(≥20X)。
项目周期:SNP检测周期为30天;全部变异检测周期为40天,个性化分析时间需根据项目实际情况进行评估。
变异检测变异分析InDel检测、注释及统计SV检测、注释及统计CNV检测、注释及统计转座子检测、注释及统计该研究对43个牛关键个体(代表Fleckvieh种群68%多态性)进行二代测序,测序深度为4.17-24.98X,平均深度7.46X。
与参考基因组比对,检测出1700万个多态性位点,91,733个变异在18,444个基因编码区中,且46%为非同义突变。
非同义突变中,575个变异可能与转录提前终止相关,3个变异出现在OMIA数据库中并与特定表型相关。
结果表明,通过对能代表种群主要多态性的关键个体进行低、中深度测序,能够高效、准确地获得该物种大量多态性信息。
案例二 牛种群关键个体中低深度测序评估基因组多态性[2]图1 Red-1水稻基因组的变异特征图3 变异注释[1] Cheng Z, Lin J, Lin T, et al . Genome-wide analysis of radiation-induced mutations in rice (Oryza sativa Molecular BioSystems, 2014, 10(4): 795-805.[2] Jansen S, Aigner B, Pausch H, et al . Assessment of the genomic variation in a cattle population by re-sequencing of key animals at low to medium coverage [J]. BMC genomics, 2013, 14(1): 446.图2 SNPs、Indels 和SVs三种变异类型相关基因的WEGO聚类限制性内切酶酶切HiSeq PE150测序数据质控与参考基因组比对Tag聚类、局部组装SNP检测及统计参考基因组已知参考基因组未知利用RAD-seq或GBS技术对某一物种个体或群体的基因组进行测序及差异分析,获得SNP遗传多态性信息,开发分子标记,建立遗传多态性数据库,为分子遗传育种研究、揭示进化关系、功能基因挖掘等奠定基础。
简化基因组技术不受参考基因组限制,基于SNPs的分子标记技术性价比高、稳定性好,在基因组中分布广泛,特别适合大样本量的分析。
诺禾致源具有稳定的简化基因组实验和分析流程,以及严格的质量控制标准,检测的变异准确性好、验证率高。
变异检测(基于简化基因组测序)技术路线技术参数基因组DNARAD文库构建GBS文库构建变异分析SNP检测、注释及统计变异分析InDel检测、注释及统计简化基因组技术RAD-seq GBSDNA样品量建库测序深度参考基因组周期≥3 μg EcoR I酶切+随机打断组装样本≥5X,比对样本≥1X ≥2 μgMse I、EcoR I、Nla III、Hae III等单酶或组合酶切Tag数≥10万,平均8X/Tag有无参考基因组均可标准分析为40天,个性化分析需根据项目实际情况进行评估变异检测图1 301株精选大豆品系的GBS分型结果案例二 GBS技术对大豆育种群体进行基因分型和基因组预测[2]随着基因分型技术的进步,如GBS技术,使分子遗传育种的周期和费用大幅下降。
该研究评估了GBS技术在大豆育种中的应用前景,结果表明GBS技术在大豆育种选择中具有重要潜力(图1)。
参考文献[1] Bus A, Hecht J, Huettel B, et al. High-throughput polymorphism detection and genotyping in Brassica napus using next- generation RAD sequencing [J]. BMC genomics, 2012, 13(1): 281.[2] Jarquín D, Kocak K, Posadas L, et al. Genotyping by sequencing for genomic prediction in a soybean breeding population [J]. BMC Genomics, 2014, 15:740.基因组DNAHiSeq PE150测序与参考基因组比对SNP 频率差异分析目标性状相关区域定位候选基因功能注释350 bp小片段文库数据质控SNP检测及注释针对研究的目标性状,选择表型极端差异的亲本构建家系。
利用混池分组分析法(Bulk Segregant Analysis, BSA),对该家系目标性状表型极端的子代分别混合成的两个样本池进行测序,同时对亲本进行测序,检测与性状相关联的位点并注释,研究基因控制目标性状的机制。
诺禾致源已完成多种动植物BSA性状定位项目,具有丰富的项目经验。
BSA性状定位技术路线技术参数DNA样品量:≥3 μg测序策略:亲本:10X/个 子代:20X/池项目周期:标准分析时间40天,个性化分析需根据项目实际情况进行评估适用范围: 1) 单倍体、二倍体或多倍体物种(有参考基因组序列) 2) 家系群体(F 2、RILs、DH等家系群体)BSA 性状定位本研究采用BSA混样策略,对10株极端性状的样品(F 2子代群体)混合的DNA池,及其亲本进行基因组重测序,通过全基因组扫描SNP,分析频率差异,检测F 2群体早花性状的QTL,找到了一个位于早花QTL 群体,SSR标记构建的遗传图谱进行QTL定位。
两种策略结合,将Ef1.1案例二 QTL-seq定位黄瓜早花重要农艺性状[2]图1 水稻耐盐性突变基因的鉴定图2 黄瓜早花性状在亲本和F 1代的表现及子代极端池SNP-index差异参考文献[1] Takagi H, Tamiru M, Abe A, et al . MutMap accelerates breeding of a salt-tolerant rice cultivar [J]. Nature biotechnology, 2015.[2] Lu H, Lin T, Klein J, et al . QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber [J Theoretical and applied genetics, 2014, 127(7): 1491-1499.]350 bp小片段文库功能基因挖掘是对地方驯化品种中不同品种或品系及其野生型,采用对DNA样品混池后建库,或者对所有单个个体DNA样品进行建库的方法,通过全基因组重测序,运用生物信息学方法全基因组范围内扫描变异位点,检测驯化性状相关的基因区域及其功能基因。
诺禾致源拥有资深的信息分析团队,项目经验丰富,功能基因挖掘为我公司首推产品。
功能基因挖掘技术路线基因组DNADNA样品混池DNA样品不混池HiSeq PE150测序数据质控与参考基因组比对SNP检测及注释选择消除分析驯化性状相关区域定位候选基因功能注释技术参数建库方式适用范围周期建库策略多个个体DNA混池测序单个个体建库自然群体DNA样品量≥3 μg DNA池样本数目≥20个/池具有性状差异的有参个体每个群体≥8个个体60天20X/池测序策略5X/个功能基因挖掘图1 候选驯化区域的选择性清除分析案例二 基于全基因组重测序探究狗对淀粉类食物的适应性的关键基因[2]狗被认为是狼驯化的产物,两者在行为和形态上存在极大差异。
研究者应用二代测序技术,对分布于世界各地的7个品种的狼(12只)和14个品种的狗(60只)采用混合测序(Pooling)方式进行了全基因组重测序,共混合6个池,平均每个池测序6X。
通过选择消除分析方法,挖掘到狗驯化过程中受到人工选择,导致现代狗行为和形态与狼存在差异图2 37个候选驯化区域的选择性清除分析[1] Li , Tian , Yeung , et al. Whole-genome sequencing of Berkshire (European native pig) provides insights into its origin and domestication [J]. Nature Scientifc Reports, 2014, 4(4).[2] Axelsson E, Ratnakumar A, Arendt M L, et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet [J]. Nature, 2013, 495(7441): 360-364.基于全基因组重测序技术对已有参考基因组序列的物种进行个体或群体的全基因组测序,利用高性能计算平台和生物信息学方法,检测单核苷酸多态性位点(SNP),并计算多态性标记间的遗传连锁距离,绘制高密度的遗传图谱。