分布容积血浆蛋白结合率
- 格式:ppt
- 大小:223.00 KB
- 文档页数:34

药代动力学参数回顾引言药代动力学是研究药物在人体内的吸收、分布、代谢和排泄过程,旨在了解药物在体内的动力学特性。
药代动力学参数是对药物在体内行为进行量化和描述的指标,可以用来预测给药方案、评估药物疗效以及调整药物剂量等。
常见药代动力学参数1. 生物利用度(Bioavailability)生物利用度是指口服给药后经胃肠道吸收到达整个体系的药物比例。
通常以F表示,计算公式为:F = (AUC口服 / AUC静脉注射)× 100%。
生物利用度越高,药物在体内的有效浓度越大,对患者的治疗效果越好。
2. 血浆蛋白结合率(Plasma Protein Binding)血浆蛋白结合率是指药物与血浆蛋白结合形成复合物的比例。
蛋白结合率越高,药物与蛋白结合的数量越多,有效游离于血浆中的药物浓度越低,可能影响药物的疗效和副作用。
3. 分布容积(Volume of Distribution)分布容积是描述药物分布范围大小的参数,表示药物在体内分布的广度。
计算公式为:Vd = 药物总量 / C0,其中C0为给药后血浆药物浓度。
分布容积越大,说明药物在组织中的浓度较高,对组织起作用的可能性也较大。
4. 消除半衰期(Elimination Half-life)消除半衰期是指体内药物浓度下降到原来半数所需的时间。
消除速率常用半衰期来表示,可根据消除速率计算出来。
消除半衰期越长,说明药物在体内的代谢和排泄速度较慢,剂量间隔较长,患者需要较少次数的给药。
5. 清除率(Clearance)清除率是描述从体内完全清除药物所需的速度。
清除率可以通过总药物量除以AUC(面积下曲线)得到。
清除率越大,说明药物在体内的代谢和排泄速度越快,患者可能需要更频繁的给药。
结论药代动力学参数对于药物的合理使用和治疗效果的评估起着至关重要的作用。
了解和评估这些参数可以帮助医生和药师更好地选择和调整药物方案,从而确保药物的安全有效使用。
以上是对常见药代动力学参数的回顾,希望能够对读者有所帮助。

血液净化技术应用于临床治疗急慢性肾功能衰竭已有近半个世纪,而在对危重患者如急性肾衰(ARF)的治疗中,连续性肾脏替代治疗(CRRT)较传统的间歇性血液透析具有更大的优势,其临床应用范围也逐渐扩大,从传统的肾脏替代向肾脏支持发展,参与多学科危急重症抢救。
无论CRF还是ARF患者,通常应用多种药物治疗,药物在这些患者的应用中须根据其残余肾功能调整,而同时,血液净化又改变了这些患者体内药物代谢情况,尤其在危重患者中,如未能及时考虑此因素,调整用药方案,后果可能不堪设想。
一)从以下三方面评估血液净化患者的药动学。
一、药物性质1、肾脏在药物清除中所占比重:药物在机体中的总体清除率是机体各器官系统清除药物能力的总和,包括肝脏、肾脏、以及其他代谢途径。
如果药物主要通过肾脏清除,则CRRT也通常可清除部分,当体外清除/总体清除≥25~30%,就需要药物剂量调整。
2、蛋白结合率:游离的药物具有生物活性并可以被滤过清除,血浆蛋白结合率高的药物则(如洋地黄毒甙、华法林等)很难被CRRT清除。
蛋白结合率可被多种因素影响,理论数值可能与实际情况有一定差异。
3、分子量:小分子易以弥散方式通过透析膜孔,药物清除与分子大小成反比,大分子常以对流通过,除非其分子量超过膜孔大小,否则清除与超滤率相关。
多数药物的分子量小于500Da,很少大于1500Da。
高通量膜及延长透析时间可改善较大分子清除。
4、分布容积(Vd):代表药物在体内组织分布的广泛程度。
Vd高代表药物组织结合率高,清除率就低。
重症患者Vd可和理论值有很大差异,而且存在个体间差异。
药物Vd≤1L/kg 易清除,≥2L/kg难以清除。
高流量IHD可将较高Vd的药物迅速从血浆中清除,降低血药浓度,但是一次透析清除只是体内药物一小部分,在两次透析之间,血药浓度会迅速回升。
CRRT持续缓慢地清除高Vd药物,此过程中药物可从组织到血浆进行重分布,其在血浆浓度的改变很小。
5、药物电荷:滤过膜常吸附阴离子,带负电荷,带正电荷的药物滤过减少,而带负电荷的药物滤过率增加。



8个常用药代动力学参数药代动力学是指药物在体内的吸收、分布、代谢和排泄过程,对药物的药效和药物副作用都有重要影响。
了解药代动力学参数是合理用药的基础,下面将介绍8个常用的药代动力学参数。
1. 生物利用度(Bioavailability):生物利用度是指药物经口给药后在体内可用形式的百分比。
一般用血药浓度曲线下面积(AUC)来表示,生物利用度越高,则药物吸收效果越好。
2. 最大血药浓度(Cmax):最大血药浓度是药物给药后在体内达到的最高血药浓度。
测量Cmax可用来评估药物的吸收效果,也可以用来确定药物的最大耐受剂量。
3. 药物分布容积(Volume of Distribution,Vd):药物分布容积是指在体内分布药物的组织总容积,计算公式为药物总体内含量除以血药浓度。
Vd反映了药物在体内分布的广泛程度,可以用于评估药物的组织分布特性。
4. 清除率(Clearance,CL):清除率是指单位时间内从体内清除药物的能力。
通常用血浆清除率(plasma clearance)来表示,血浆清除率越大则药物从体内清除速度越快。
5. 半衰期(Half-life,t1/2):半衰期是指药物在体内降解清除一半所需的时间。
半衰期可以反映药物的排除速度,药物的给药间隔时间通常与其半衰期有关。
6. 生物转化率(First-pass metabolism):生物转化率是指药物在通过肝脏或其他代谢器官之前,在消化道或肝脏内发生的首次通过转化的百分比。
生物转化率通常影响药物的生物利用度。
7. 蛋白结合率(Protein Binding):药物分子与血浆中的蛋白质结合形成药物蛋白复合物,只有未结合的药物才能发挥药效。
蛋白结合率表示结合在蛋白质上的药物所占的百分比,蛋白结合率高表示药物与蛋白质结合紧密,对于药物的分布和消除有重要影响。
8. 药物代谢饱和点(Saturation Kinetics):药物在体内代谢是一个饱和过程,当药物被代谢酶饱和时,药物在体内代谢速率不再随药物浓度的增加而增加,这种饱和状态称为代谢饱和。

前言依据游离药物假说的基本内容,只有游离态的药物才能发挥药物浓度基本已经形成共识,在药物发现阶段我们是否可以通过结构修饰增加药物的游离比例(fraction of unbound, fu)来达到增加游离药物浓度的目标?理解该问题有利于早期化合物的高效筛选,避免陷入迷宫。
常规情况下,从DMPK的角度来看,血浆蛋白结合率(plasma protein binding, PPB)不适合作为主要的筛选指标,因为同时有理论认为只有游离态药物才能被清除。
下文将就这一问题从理论分析和实验数据两个角度进行阐述,以期助于大家理解PPB的合理运用。
1 理论分析基于游离药物才能发挥药效的理论,我们可以已基本确定我们药物发现阶段的最终目标是拿到有合适的Cu或AUCu的化合物。
一般Cu和AUCu 用下式表示:Cu=fu*CAUCu=AUC*fu,大多药物的PK/PD关系为AUCu驱动,后文我们将以口服药物的AUCu的计算为例进行解读。
下式为AUC和AUCu计算方法,。
AUC=Dose*F/CLAUCu= fu*Dose*F/CL同时基于只有游离药物浓度才能被清除的理论,假设药物的清除主要依赖肝脏代谢,依据well-stirred模型,假设药物吸收良好,吸收比例分数Fa=1,无肠道代谢,则CL和F则可换算为下式,。
CL=Q*fu*Clint/(Q+fu*Clint),F=1-ER=1-CL/Q= Q /(Q+fu*Clint)此处Q为肝血流速率,Clint为固有清除率;将CL和F的换算值带入方程AUC=Dose*F/CL和AUCu= fu*Dose*F/CL中,即可得:AUC=Dose/fu/ClintAUCu= Dose/Clint到这里就可以发现虽然AUC与fu有着相反的关系,但是AUCu与fu 半毛钱关系没有,仅与Clint有关,Clint反映的正是化合物的代谢稳定性,这也就是代谢稳定性能够经常作为T1级别的筛选指标的原因。


药物血浆蛋白结合率的临床意义相比于半衰期、达峰时间、生物利用度等「明星」指标,药物的血浆蛋白结合率往往并不起眼;很多人即便注意到这一指标,也难以准确解读其意义,甚至仅仅将其视为一个空泛的数字。
然而,如果对血浆蛋白结合率这一指标缺乏足够的认识,某些临床情况下则可能引发严重的后果。
以下对该指标进行简要介绍——临床意义简言之,当药物与血浆蛋白结合时,这一部分药物无法发挥效应,无法穿越血脑屏障,也无法被清除。
当游离态药物被清除时,原先与蛋白结合的药物可以解离出来,继续发挥药理学效应。
换言之,与血浆蛋白结合的药物发挥了类似「贮水池」的作用,在需要的时候可为机体提供游离态药物。
结合态药物、游离态药物与药物清除率之间存在动态平衡。
假设一种药物的蛋白结合率为99%,只有1%处于游离态,那么一旦与蛋白结合的药物比例下降至98%,处于游离态的药物比例则升高至2%。
虽然仅仅增加了1%,但已经相当于游离态药物浓度翻番,可能造成一系列后果。
与药物结合的蛋白主要包括白蛋白、α-1-酸性糖蛋白(AAG)及脂蛋白。
白蛋白占血浆全部蛋白的60%,很多疾病可导致低蛋白血症,最常见的包括营养不良、恶病质、应激、外伤、烧伤、妊娠及糖尿病等。
一旦可供结合的白蛋白减少,「贮水池」变小,处于游离态的药物水平则相应升高,而药物剂量也可能需要酌情调整。
相比于白蛋白,AAG在血浆中所占比重较低,临床意义也相对较弱。
然而值得注意的是,AAG水平在通常状况下并不会降低,但在创伤、炎症、急性心梗等情况下可能升高,造成药物与蛋白结合的增加及游离态药物水平的降低,理论上可能导致疗效的下降。
脂蛋白主要包括高密度脂蛋白(HDL)、低密度脂蛋白(LDL)等,环孢霉素、他克莫司及丙泊酚均可与脂蛋白结合。
另外,还有一些药物位点可以与红细胞血红蛋白及质膜结合,包括巴比妥酸盐、氯丙嗪、丙米嗪及苯妥英,此种情况也属于与血浆蛋白结合。
表1 蛋白结合率>90%的部分临床常用药(Eichel EA. 2018)表1列举了蛋白结合率>90%的部分临床常用药。


血浆蛋白结合率对药物作用的影响
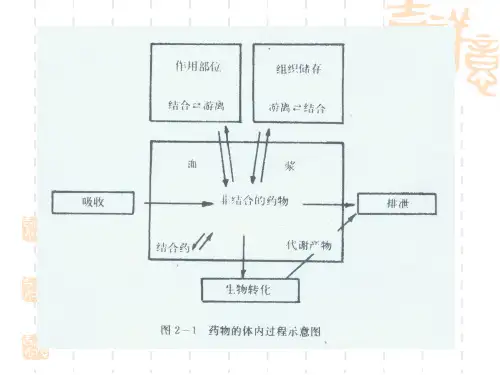
血浆蛋白结合率:药物进入血液后与血浆蛋白结合的量占血液总药量的比例。
各种药物以一定的比率与血浆蛋白结合,在血浆中常同时存在结合型与游离型。
而只有游离型药物才具有药物活性。
药物与血浆蛋白结合成为结合型药物,暂时失去药理活性,并“储存”于血液中,起到药库的作用。
对于药物作用及其维持时间长短有重要意义。
结合分为可逆性、饱和性、非特异性、竞争性。
药物与血浆蛋白的结合影响药物在体内的分布、转运速度以及作用强度和消除速率。
一般蛋白结合率高的药物体内消除慢,作用维持时间长,药效平稳。
结合率低的药物体内消除快,同时作用时间短,药效有很大的波动。
药物内源性性化合物也可在血浆蛋白结合部位发生竞争性置换作用,两种以上的药物联用时,可相互竞争血浆蛋白的结合部位,结合力强的药物能从蛋白结合部位上取代结合力弱的药物,使后者游离型数量增加,导致药效和毒性反应亦增强。
其影响程度可因后者在体内的分布容积不同而异。
一般只有血浆蛋白结合率高,分布容积小,消除慢以及治疗指数低的药物在临床上的这种相互作用才有意义。
临床药物效应动力学名词解释药物效应动力学是研究药物在体内的吸收、分布、代谢和排泄等过程以及与药物剂量、时间、用药途径等因素的关系,从而揭示药物在体内的作用机制和药效的变化规律。
在临床应用中,了解药物效应动力学是非常重要的,因为它可以指导临床医师合理用药,避免药物不良反应和药物相互作用等问题。
以下是一些常见的药物效应动力学名词解释。
一、药物吸收动力学1. 生物利用度(Bioavailability)生物利用度是指药物在体内吸收的程度和速度,通常用药物口服后的血浆药物浓度曲线下面积(AUC)来衡量。
药物的生物利用度受到许多因素的影响,如药物溶解度、肠道pH值、肠道蠕动、肠道酶活性等。
2. 最大血浆药物浓度(Cmax)最大血浆药物浓度是指药物在吸收后达到的最高血浆浓度,通常发生在给药后的1-2小时内。
Cmax反映了药物的吸收速度和程度,是评价药物生物利用度的一个重要指标。
3. 时间至最大血浆药物浓度(Tmax)时间至最大血浆药物浓度是指药物在吸收后达到最大血浆浓度所需要的时间。
Tmax通常发生在给药后的1-2小时内,但也有例外,如缓释剂型的药物可能需要更长时间才能达到最大血浆浓度。
二、药物分布动力学1. 血浆蛋白结合率(Plasma protein binding)药物在体内往往与血浆中的蛋白质结合,从而影响药物的分布和代谢。
血浆蛋白结合率是指药物与血浆蛋白结合的比例,通常用百分比来表示。
血浆蛋白结合率高的药物往往分布范围较小,药效持续时间较短。
2. 分布容积(Volume of distribution)分布容积是指药物在体内分布的范围,通常用药物总量除以血浆药物浓度来计算。
分布容积大的药物往往分布范围广,药物作用时间较长。
三、药物代谢动力学1. 代谢酶(Metabolic enzymes)药物在体内的代谢主要由肝脏中的代谢酶完成,如细胞色素P450酶(CYP450)等。
代谢酶的种类和活性不同,会影响药物的代谢速度和药效。
药动学的参数药物动力学是研究药物在体内吸收、分布、代谢和排泄过程的科学。
正确认识和掌握药物动力学的参数对于合理用药、临床治疗以及新药开发都具有重要意义。
本文将从吸收、分布、代谢和排泄四个方面分别介绍药物动力学的主要参数。
一、吸收参数1. 生物利用度(bioavailability):药物经口给药后进入体内的总吸收量与直接注射的总吸收量的比值。
一般通过体内外药物浓度随时间的变化曲线来计算。
2. 最大血浓度(Cmax):药物在给药之后血浆中达到的最高浓度。
该参数反映了药物吸收的速度和程度。
3. 时间到达峰浓度(Tmax):最大血浓度出现所需要的时间。
4. 血药浓度-时间曲线(AUC):药物在体内的总曝露量,通过血浆药物浓度随时间的变化曲线下的面积来计算。
AUC值越大,表示总体内曝露量越大。
二、分布参数1. 分布容积(Vd):反映药物在体内分布的广泛程度。
Vd值越大,说明药物在体内的分布范围越广。
2. 血浆蛋白结合率(protein binding):部分药物在体内主要以结合蛋白的形式存在,该参数反映了药物与蛋白质结合的程度。
3. 大脑-血浆分布系数(BBB):反映了药物能否越过血脑屏障进入脑组织,对于中枢神经系统药物很重要。
三、代谢参数1. 代谢速率(metabolic clearance):单位时间内从血浆中清除药物的速率。
代谢速率越大,药物代谢越快。
2. 代谢半衰期(t1/2):药物浓度下降到初始浓度的一半所需要的时间。
代谢半衰期可间接反映药物在体内停留的时间。
四、排泄参数1. 肾脏清除率(renal clearance):药物在单位时间内通过肾脏清除的速率。
2. 肝脏清除率(hepatic clearance):药物在单位时间内通过肝脏清除的速率。
药物动力学参数反映了药物在体内的代谢、分布、排泄等过程。
了解这些参数有助于合理用药,指导临床用药,也有助于评价新药的临床前药代动力学特性。
对于药物研究、开发和用药安全有着重要的意义。
平衡透析法检测血浆蛋白结合率原理及注意事项药物血浆蛋白结合率(Plasma Protein Binding, PPB)即血液中与蛋白结合的药物占总药量的百分数,可以反映药物与血浆蛋白结合的程度。
药物血浆蛋白结合率是药物在动物体内重要的药理学参数之一,影响着药物体内游离浓度进而影响药物的分布与排泄。
药物在进入血液后与血浆蛋白会有不同程度结合,血液中游离药物的比例会影响其在体内吸收、分布、代谢和排泄的过程,与血浆蛋白结合的药物可能会有更长的半衰期和更慢的清除速率,因此测定血浆蛋白结合率对于理解化合物的活性以及组织分布有很重要的参考意义。
1 结合蛋白类型血浆蛋白主要作为载体负责各类外源性和内源性分子在循环系统内的转运。
其中参与小分子药物结合的主要有白蛋白、α-1-酸性糖蛋白(AAG)、球蛋白和脂蛋白,主要以白蛋白和α-1-酸性糖蛋白(AAG)为主。
药物与蛋白的结合强弱主要与药物-蛋白亲和力、蛋白丰度相关。
2 血浆蛋白结合率常用测定方法目前常用于药物血浆蛋白结合率测定的常用方法包括平衡透析法(Equilibrium Dialysis),超滤法(Utrafiltration Method),超速离心法(Ultracentrifugation Method),凝胶过滤法(Gel filtration)等。
其中,平衡透析法是基于药物结合的平衡原理来测定药物游离浓度最常用的方法,也是研究药物血浆蛋白结合率的经典方法。
超滤法使用滤膜分立自由的药物和结合蛋白的药物,时间短,但确定点是药物容易结合到超滤过程使用的器械上,从而影响检测结果。
平衡透析法虽然比超滤法耗时多一些,但它使用teflon镀层的平衡装置,能大大减少药物的非特异性结合,得到更精确的结果。
目前,平衡透析法已有96孔板的高通量测试形式,可以实现同时测试大量候选药物的蛋白结合率。
平衡透析法常用种属血浆:大鼠血浆,小鼠血浆,比格犬血浆,猴血浆,人血浆分析方法:HPLC,LC-MS/MS,GC-MS等各种药物以一定的比率与血浆蛋白结合之后,在血浆中会同时存在结合型与游离型两种类型,游离性药物具有药物活性。
评价药物分布能力的参数一、引言药物分布是药物在体内分布的过程,它直接影响药物的疗效和毒性。
因此,评价药物分布能力的参数对于临床用药和新药研发具有重要意义。
二、血浆蛋白结合率血浆蛋白结合率是指药物与血浆蛋白结合的程度,它决定了游离态药物的浓度和有效性。
通常来说,结合率越高,游离态药物的浓度越低,对机体产生的作用也越小。
因此,血浆蛋白结合率是评价药物分布能力的一个重要参数。
三、体内分布容积体内分布容积是指单位时间内将所有游离态药物从血液中排除所需要的总体积。
它反映了游离态药物在机体内部分散到多少组织中,并且是影响靶组织中药物浓度的主要因素之一。
因此,在评价药物分布能力时,需要考虑其体内分布容积。
四、组织亲和力组织亲和力是指游离态药物与不同组织间的亲和性,它影响药物在不同组织中的分布。
通常来说,药物在靶组织中的浓度越高,其作用越强。
因此,在评价药物分布能力时,需要考虑其组织亲和力。
五、血脑屏障透过性血脑屏障是指由神经元、星形胶质细胞和毛细血管内皮细胞构成的一种生理屏障,它限制了大多数药物进入中枢神经系统。
因此,在评价药物分布能力时,需要考虑其血脑屏障透过性。
六、药物代谢和排泄药物代谢和排泄是指机体对于药物的代谢和排出过程。
它决定了体内游离态药物浓度的降低速度。
因此,在评价药物分布能力时,需要考虑其代谢和排泄速率。
七、结论评价药物分布能力的参数包括血浆蛋白结合率、体内分布容积、组织亲和力、血脑屏障透过性以及药物代谢和排泄等方面。
这些参数相互作用,共同影响药物在体内的分布和疗效。
因此,在临床用药和新药研发中,需要综合考虑这些参数,并根据具体情况进行合理的选择和调整。
生物药剂学模拟题[A型题]1 药物的灭活和消除速度主要决定A起效的快慢B作用持续时间C最大效应D后遗效应的大小E不良反应的大小正确答案:B2 药物消除半衰期(t1/2)指的是下列哪一条A吸收一半所需要的时间B进入血液循环所需要的时间C与血浆蛋白结合一半所需要的时间D血药浓度消失一半所需要的时间E药效下降一半所需要的时间正确答案:D3 关于生物利用度的描述,哪一条是正确的A所有制剂,必须进行生物利用度检查B生物利用度越高越好C生物利用度越低越好D生物利用度应相对固定,过大或过小均不利于医疗应用E生物利用度与疗效无关正确答案:D4 下列关于药物在体内半衰期的叙述哪个是正确的A随血药浓度的下降而缩短B随血药浓度的下降而延长C在一定剂量范围内固定不变,与血药浓度高低无关D在任何剂量下,固定不变E与首次服用的剂量有关正确答案:C5 为迅速达到血浆峰值,可采取下列哪一个措施A每次用药量加倍B缩短给药间隔时间C首次剂量加倍,而后按其原来的间隔时间给予原剂量D延长给药间隔时间E每次用药量减半正确答案:C6 设人体血流量为5L,静脉注射某药物500mg,立即测出血药浓度为1mg/ml,按一室分配计算,其表观分布容积为多少A 0.5LB 7.5LC 10LD 25LE 50L正确答案:A7 作为一级吸收过程输入的给药途径下列哪项是正确的A多次静脉注射B静脉注射C肌内注射、口服、直肠、皮下注射等凡属非血管给药途径输入者D以上都不对E以上都对正确答案:C8 Wanger-Nelson法(简称W-N法)是一个非常有名的方法,它主要是用来计算下列哪一个模型参数A吸收速度常数KaB达峰时间C达峰浓度D分布容积E总清除率正确答案:A9 非线性药物动力学中两个最基本而重要的参数是哪一项A t1/2和KB V和ClC Tm和CmD Km和VmE K12和K21正确答案:D10 下列哪一项是混杂参数A α、β、A、BB K10、K12、K21C Ka、K、α、βD t1/2、Cl、A、BE Cl、V、α、β正确答案:A11 关于平均稳态血药浓度下列哪一项的叙述是正确的A是(C∞)max与(C∞)min的算术平均值B在一个剂量间隔内(τ时间内)血药浓度曲线下面的面积除以间隔时间τ所得的商C是(C∞)max与(C∞)min的几何平均值D是(C∞)max除以2所得的商E以上叙述都不对正确答案:B12 拟定给药方案的设计时,要调整剂量主要调节以下哪一项A (C∞)max和(C∞)minB t1/2和KC V和ClD X0和τE以上都可以正确答案:D13 就下图回答下各项哪一项是正确的A单室静注图形B双室静注图形C单室尿药图形D单室口服图形E以上都不是正确答案:B14 下列哪一个方程式符合所给图形A C=KaFX0/[V(Ka-K)](e-Kt-e-Kat)B C=C0e-KtC C=Ae-Kat+Be-βtD C=X0/(KVτ)(1-e-Kt)E以上都不对正确答案:A15 下列哪个公式是单室模型静脉注尿药速度法A log(Xu∞-Xu)=-Kt/2.303+logXu∞B logC=-Kt/2.303+logC0C logdXu/dt=-Kt/2.303+logKeX0D log△X0/△t=-Kt/2.303+logKeKaFX0/(Ka-K)E A和C都对正确答案:C16 在新生儿时期,许多药物的半衰期延长,这是因为A较高的蛋白结合率B微粒体酶的诱发C药物吸收很完全D酶系统发育不全E阻止药物分布全身的屏障发育不全正确答案:D17 线性药物动力学的药物生物半衰期的重要特点是A主要取决于开始浓度B与首次剂量有关C与给药途径有关D与开始浓度或剂量及给药途径有关E与给药间隔有关正确答案:D18 生物等效性的哪一种说法是正确的A两种产品在吸收的速度上没有差别B两种产品在吸收程度上没有差别C两种产品在吸收程度与速度上没有差别D在相同实验条件下,相同剂量的药剂等效产品,它们吸收速度与程度没有显著差别E两种产品在消除时间上没有差别正确答案:D19 消除速度常数的单位是A时间B毫克/时间C毫升/时间D升E时间的倒数正确答案:E20 今有A、B两种不同药物,给一位患者静脉注射,测得其剂量与半衰期数据如下,请问下列叙述哪一项是正确的剂量(mg) 药物A的t1/2(h) 药物B的t1/2(h)40 10 3.4760 15 3.4780 20 3.47A说明药物B是以零级过程消除B两种药物都以一级过程消除C药物B是以一级过程消除D药物A是以一级过程消除E A、B都是以零级过程消除正确答案:C21 如果处方中选用口服硝酸甘油,就应选用较大剂量,因为A胃肠道吸收差B在肠中水解C与血浆蛋白结合率高D首过效应明显E肠道细菌分解正确答案:D22 用于比较同一药物两种剂型的生物等效性的药代动力学参数是A AUC、Vd和CmaxB AUC、Vd和TmaxC AUC、Vd和t1/2D AUC、Cmax和TmaxE Cmax、Tmax和t1/2正确答案:D23 红霉素的生物有效性可因下述哪种因素而明显增加A缓释片B肠溶衣C薄膜包衣片D使用红霉素硬脂酸盐E增加颗粒大小正确答案:B24 一位病人在饥饿与饭后的条件下各服用某非巴比妥催眠药,其血药浓度变化如图所示,据此下列哪种答案是正确的A饥饿时药物消除较快B食物可减少药物吸收速率C应告诉病人睡前与食物一起服用D药物作用的发生不受食物的影响E由于食物存在,药物血浆峰度明显降低正确答案:B25红霉素硬脂酸盐包衣片由健康志愿者服用,下图为5种不同条件下的血药浓度----时间曲线,从图上看,药剂师可以正确的判断≠对五种服药方式,其红霉素的血浆峰浓度出现于同一时刻≡当服红霉素盐时同服250ml水可得到最高血浆红霉素水平≈每种处理都得到血药浓度曲线下相等面积A只包括≠B只包括≈C只包括≠和≡D只包括≡和≈E ≠、≡和≈全部包括正确答案:C26 若罗红霉素的剂型拟以片剂改成注射剂,其剂量应A增加,因为生物有效性降低B增加,因为肝肠循环减低C减少,因为生物有效性更大D减少,因为组织分布更多E维持不变正确答案:C27 下面是关于用线性一室模型解释药物体内动态分布容积的叙述,哪个组合是正确的≠分布容积是药物在体内转运的组织真实容积≡如果药物向组织转运过多,则分布容积的值越小≈如果药物向组织转运较多,则分布容积的值越大⋯分布容积可用快速静脉给药时的剂量(D)与血药浓度(C0)的乘积(D*C0)来计算A ≠∣B ≡⋯C ≡∣D ≈⋯E ≈∣正确答案:D28 下面关于氨基糖苷类抗生素的叙述中,哪种组合是正确的≠因为在消化道吸收很少,所以没有口服制剂≡大部分以原型药物从肾排泄≈与血浆蛋白的结合较少⋯主要的毒性为肾毒性和听觉器官毒性≠≡≈⋯A 正误正误B 正正误误C 误误正正D 误正误正E 误正正正正确答案:E29 药物A的血浆蛋白结合率(fu)为0.02,恒速滴注达稳态后的血中药物浓度为2μg/ml。