
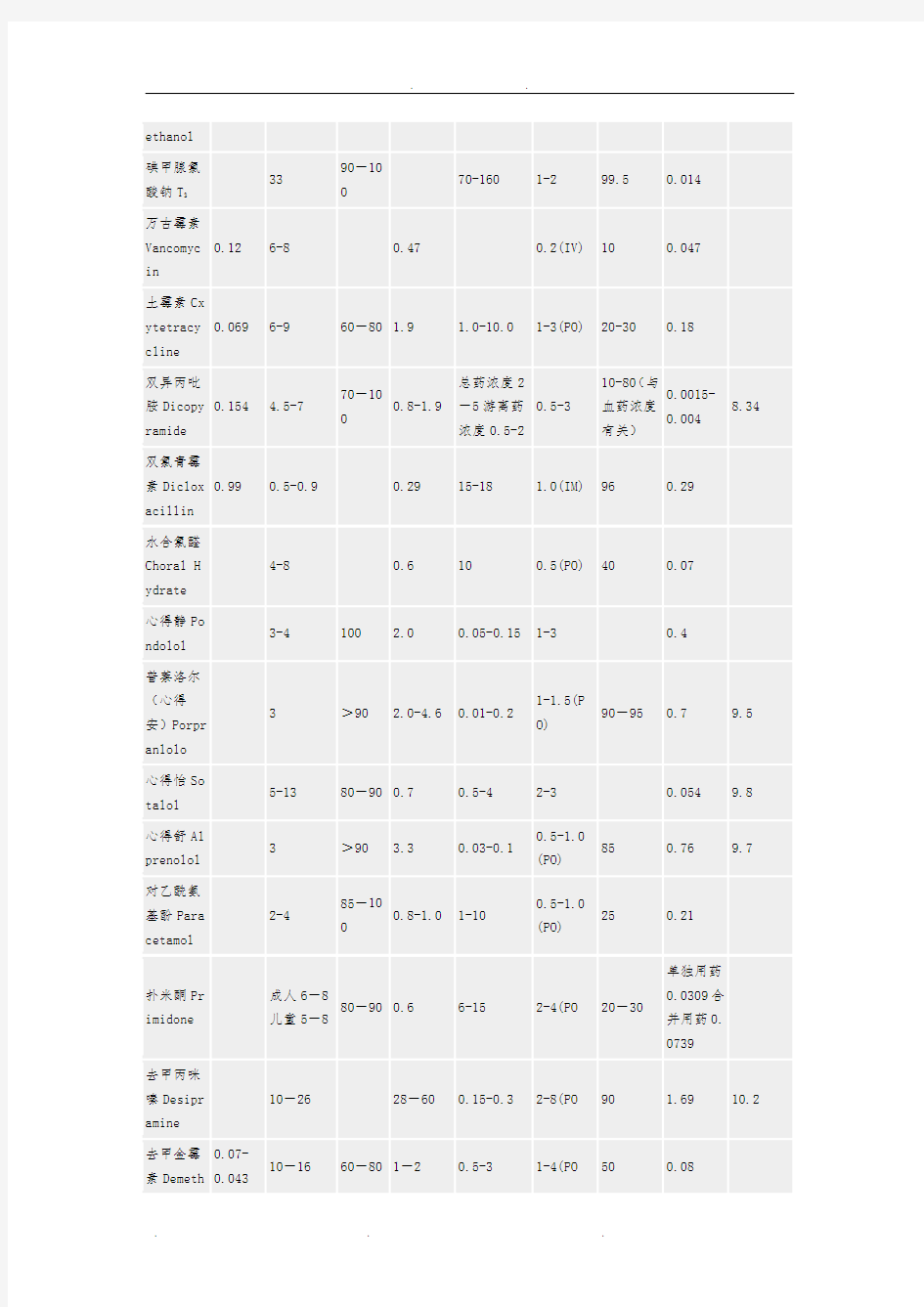
八、常用药物的药代动力学参参考表
药物名称消除速
率常数
k或β
(hr-1)
半衰期tl
/2或tl/2
β
口服吸
收(%)
表现分布
容积vb(m
g/L)
有效血浓度
围(mg/L)
峰时tm(h
r)
蛋白结合
率BP(%)
消除率Cl
s[L/(hr.
kg)]
解离指数
pKa
乙基西梭
霉素Ncti lmicin 0.312 2.23 0.25-0.3 4-8
0.5-1.0
(IM)
0.78
乙氧萘青
霉素Nafi llin 1.39 0.5 0 0.28
0.5-1.0
(IM)
90 0.39
2.6(-COO
-)
乙胺丁醇
Ethambut
ol
0.198 3.5 30 1.87 1-10 2-4(PO 40 0.37
乙琥胺Et hosuximi de 成人50-
60儿童30
-50
100
成人0.62
儿童0.69
40-100 2-4(PO <2
成人0.01
-0.013儿
童0.016
1.3
乙酰唑胺
Acetazol amide 0.169
4.1
2.4-
5.8
>90 0.2
10-10.5
10-15
0.034
7.2
9.0
乙酰普鲁
卡因胺Ac
etylproc
ainamide
0.082 8.5 85 1.5 2-20 10 0.12
二甲胺四
环素Mino
cycline
12 90 0.43 0.5-3.0 1-3(PO 75 0.025
二性霉素
B Amphot
ericinB
0.029 18-24 <3 0.5-1.0 >90 0.025
二氮嗪Di
azoxid
21-36 100 0.2 90-95 0.005
丁胺卡那霉素Amik acin 0.283-
0.311
2-3
90-10
0.25 10-25 1.0(IM) 0-20 0.069
三氯乙醇
Trichlor
4-8 0.6 0.5(PO 40 0.07
碘甲腺氯酸钠T333
90-10
70-160 1-2 99.5 0.014
万古霉素
Vancomyc
in
0.12 6-8 0.47 0.2(IV) 10 0.047
土霉素Cx
ytetracy
cline
0.069 6-9 60-80 1.9 1.0-10.0 1-3(PO) 20-30 0.18 双异丙吡
胺Dicopy ramide 0.154 4.5-7
70-10
0.8-1.9
总药浓度2
-5游离药
浓度0.5-2
0.5-3
10-80(与
血药浓度
有关)
0.0015-
0.004
8.34
双氯青霉
素Diclox
acillin
0.99 0.5-0.9 0.29 15-18 1.0(IM) 96 0.29
水合氯醛
Choral H
ydrate
4-8 0.6 10 0.5(PO) 40 0.07
心得静Po
ndolol
3-4 100 2.0 0.05-0.15 1-3 0.4 普萘洛尔
(心得安)Porpr anlolo 3 >90 2.0-4.6 0.01-0.2
1-1.5(P
O)
90-95 0.7 9.5
心得怡So
talol
5-13 80-90 0.7 0.5-4 2-3 0.054 9.8
心得舒Al prenolol 3 >90 3.3 0.03-0.1
0.5-1.0
(PO)
85 0.76 9.7
对乙酰氨
基酚Para cetamol 2-4
85-10
0.8-1.0 1-10
0.5-1.0
(PO)
25 0.21
扑米酮Pr imidone 成人6-8
儿童5-8
80-90 0.6 6-15 2-4(PO 20-30
单独用药
0.0309合
并用药0.
0739
去甲丙咪
嗪Desipr
amine
10-26 28-60 0.15-0.3 2-8(PO 90 1.69 10.2
去甲金霉素Demeth 0.07-
0.043
10-16 60-80 1-2 0.5-3 1-4(PO 50 0.08
etracycl
ine
去甲替林
Nortrpyt
line
27 60 20 0.05-0.16 4-9(PO) 92-96 0.5
丙咪嗪Im ipramine 8-16 80-100 20-40 0.15-0.5
0.5-1.0
(PO)
95 1.73 9.5
丙基硫氧
嘧啶Prop
ylthiour
acil
2-4 80 0.2-0.4 1.0(PO) 75 0.07 8.3
丙磺舒Pr obenecid 6-18 100
0.12-0.1
8
100-200 1-5(PO) 85-95 0.01 3.4
布洛芬Br ufen 2 0.15
0.5-1.5
(PO)
99 0.052
左旋多巴
Leayodop a 3
20-30
(吸
收)
0.5-4 8.2
白霉素Le
ucomycin
0.139 4.5-5.5 2(PO) 72 苷珀酸Ca
rbenoxol ne 13-16 0.1
1-2(空腹
PO)饭后
(PO)
0.005
甲硝唑Me
tronidaz
ole
6-14 90-100 0.6-0.7 5-20 1-2(PO) 5-20 0.045
可待因Co
deine
0.231 2-4 50 3.45 0.025 1.0(PO) 0.8 8.2 戊巴比妥
Pentobar bital 21-42 70-80 0.7 1-4
初次用药
6-18长
期用药37
60-70 0.015 8.1
卡马西平Carbamaz epine 成人10-
30儿童8
-19
90-100 0.8-1.4 4-12
1.0-
2.0
(IM)
65-85
成人初次
用药0.01
1-0.026
儿童初次
用药0.02
8
卡那霉素Kanamyci 0.289-
0.33
2-2.5 90-95 0.25 2-8
0.5-1.0
(IM)1.0
0 0.077 7.2
n (PO) 头孢氨苄
(先锋霉素Ⅳ)Cep halexin 0.655 0.83-1.2 90-95 0.26
G(+)
0.5-3G
(-)1.0-12
15min(I
V)0.5-1.
0(IM)
10-15 0.18
5.3(-COO
H-)7.3(-
NH2)
头孢匹林
(先锋霉素Ⅶ)Cep hapirin. 1.155 0.6 90-95 0.23
G(+)
0.5-3G
(-)1.0-12
40-50 0.27
头孢娄利
定(先锋I I)Cephal oridine 0.62 1-1.5 90-95 0.25 同上同上
20
10-3
0.14
头孢噻吩
(先锋霉素I)Cep halothin 1.4 0.5-1.0 90-95 0.25 同上同上
65
60-8
0.23
头孢格来星(先锋
霉素Ⅲ)C enhalogl ycin 0.77 0.9 80-95
G(+)
0.5-3G
(-)1.0-12
15min(I
V)0.5-1.
0IM
24 4.7
头孢唑啉
(先锋霉素Ⅴ)Cep ahzolin 0.385 1.4-2.2 90-95 0.15 同上
5.0-1.0
(IM)5min
(PO)
75-85 0.058 2.3
头孢雷定
(先锋霉素Ⅵ)Cep ahzolin 0.809-
0.994
40-50min 90-95 0.25 同上
1.0(PO)5
min(IV)
5-20 0.23
头孢曲
(先锋Ⅶ)Cepha cetrile 0.462 1.5 90-95 0.27
G(+)
0.5-3G
(-)1.0-12
1.0(PO)5
min(IV)
22.34 0.125 1.97
甲状腺素
Thyroxin
e
144-168 90-95 0.1-0.2 0.03-0.04 99.95 0.067
甲氧苄胺嘧定Trim 0.07 8-12 2 0.5-12
1.5-3.3
(PO)
14 0.14 7.2
甲氧苯青
霉素Meth icillin 1.386 0.5-1 0 0.3-0.42 1- 0.5(IM)
50
35-6
0.33 2.7
甲基多巴
Methyldo pa 0.35 2 0.5 >2 4(PO) 0-20 0.17
2.2(-COO
H)10.6(-
NH)12.0
(-OH)
甲基多粘
菌素
0.154 405 0.5 1-2(IM)0.077 甲烯土霉
素Methac ycline 0.0533 10-16 60-68 0.97 1.0-10.0
1-4(PO)
-3(PO)
70 0.052
甲苯磺丁
脲Tobuta
mide
4-8 85-100 0.1-0.15 50-100 98 0.014 5.3 对基水酸
Para ami nosalicy lic acid 0.77 0.9
Sol:10
0混液:
70片:9
0.23 0.6
Na:盐0.5
-1.0酸型
3-4(PO)
65 0.18
1.8(-NH)
3.6(-COO
H)
四环素Te
tracycli
ne
0.099 6-10 60-80 1-2 1.0-10.0 1-3(PO) 25-30 0.13 8.3
地高辛Di goxin 0.017 30-50 50-93 6-10
0.0006-0.0
018
1-2(PO)
0.5-0.75
25 0.14
西梭霉素Sisomici n 0.305-
0.347
2.0-2.5
0.25-0.
3
4-8
0.75-1.0
(IM)
0-20 0.85
灰黄霉素
Griseofu lyin 8-24
40-70
(微晶
型)
1-2 0.3-1.3 2-4(PO) 0.065
华法令Wa rfarin 0.016
44
15-60
100 0.1 1-10 2-8(PO) 99 0.0019
4.8
5.0
优降糖Gl
ybenznyc
lamide
5-2 0.1 2-6(PO) 99-95 0.008
多粘菌素0.16 3-8 0.5-4.0 2 <10 8.9
xinB (IM) 多粘菌素
E(抗霉素)Polym yxin E 0.31 1.5-8 0.5
1-2
(IM)
<10 0.07 8.9
红霉素Er
ythromyc in 0.462 1.5-3 40 0.6 0.5-3
1-4
(PO)
70-90 0.185
8.8(-NH
2)
吗啡Morp hine 1.5-2.5 20 1 0.07-0.1
10-20min
(IM)
35 0.35
8.0
9.0
庆大霉素
Gentamyc in 0.35 2 <5 0.25 3-8
0.5-1
(IM)
25-30 0.087
氯,贝丁
酯(安妥
明)Clofi
brate
8-16 90-100 0.12 80-150 96 0.007
安定Diaz epam 20-50 75-100 0.2 0.1-1
1-2
(PO)
98 0.004
螺酯(安
体舒通)S pironola ctone 10min
90(吸
收)
0.05
3
(PO)
90 0.21 3.4
安替比林
Antipyri
ne
10-15 100 0.48-0.7 10 0.033 异丙肾上
腺素Isop renaline 2.5 0.5
80min
(PO)
10min(吸
入)
0.14 1.4
异烟肼Is oniazide 0.193
3.6(慢乙
酰化型)0.
7-2(快乙
90 0.6 0.5-1.0 1-2 18-30 0.12 1.8
0.63 酰化型)
(PO) 0.31 3.5
10.8
抗癫灵An
tiepilep sirin 8-15
0.15-0.
4
1-4
(PO)
90 0.016
哌替定Pe thidine 2-4 50-70 4.7 0.2-0.6 1-2(IM)
60
65-75
1.09 8.7
苄二甲胍
Bethanid ine 7-15 50-100 6
1-5
(PO)
8 0.38
呋喃妥因
Nitrofur antoin 0.7 1.0 0.71 1-2
2
(PO)
50-60 0.49 7.2
别嘌醇Al lopurine 2-8 80
2-6
(PO)
9.0
利多卡因
Lidocain e 0.46 1.5 20-50
1.6
(0.58-1.
19)
2-5
2-5min(I
V)0.5(I
M)
60-70 0.74 7.9
利福平Ri fampicin 0.25 2-5 90-95 0.6 0.5-10
1-2
(PO)
90 0.12
邻氯青霉
素Cloxac illin 1.39 0.5 50-80 0.15 7-14
1.0
(IM)
94 0.21 2.7
妥布霉素Tobramyc in 0.251-
0.347
1.5-3.5 90-100 0.25 2-10
1.0
(IM)
0-20 0.07
纳洛酮Na loxone 0.693 1 20 3
0.15-1
(IM)
2.1
阿托品At3-4 2-4 15-50 50 0.59 9.9
ropine
13-38 10 min
(IM)
1.0(PO)
0.27
阿米替林Amitript yline 8-20
32-40
65 8.8 0.05-0.3 95-98
0.44
0.17
9.4
乙酰水酸(阿司匹林)Acety
lsalicyl ic
Acid 2.0-30(mi
n)
80-100 0.15-0.2
20-100(解
热镇痛)10
0-250(抗
炎)
1-2
(PO)
50-80 0.29 3.5
环磷酰胺
Cyclopho sphamide 0.107 3-11 0.4-0.6
1.0
(PO)
10-20 0.05
青霉素Pe nicillin 1.386 0.5
15
(20-4
0)
0.25 1.5-1.0
0.25-0.5
(IM)
65 0.35 2.8
青紫霉素Lividomy cin 0.277-
0.372
2-2.5
1-2
(IM)
20-30
苯乙双胍
(降糖灵)Phenf ormin 0.231 3 50-70 5-10
100-240(ng
-/ml)
2-4
(PO)
20-25 1.73
苯巴比妥Phenobar bital 成人53-1
40儿童37
-94
>90
成人0.7
儿童0.7-
1.2
15-40
6-18
(PO)
45-50
成人0.00
32老人
0.0024儿
童0.0082
新生儿0.
0064
7.3
苯妥英钠Phenytoi n 成人18-3
0儿童12-
22
90 0.6-0.8 10-20
4
3-10
88-92 0.029 803
(PO) 苯海拉明
Diphenhy dramine 4-10 50 3-4 0.01-0.1
2-4
(PO)
98-99 0.35
9.0
7.7
苯唑青霉
素Oxacil lin 1.386 0.5-1.0 70 0.25 5-6
1.0(PO)
0.5-1.0
(IM)
90 0.23
苯磺唑酮
Sulfinpy
razine
3-5 90-100 0.15 1.0(PO) 98-99 0.026 林可霉素
Limcoyci n 0.154 4-6 40 0.35 2-10
2-4
(PO)
1-2
(IM)
70 0.049
7.6(-NH
2)
金刚烷胺
Amamtadi n 0.315 22
90-10
6
1-4
(PO)
0.19
肼苯达嗪
Hydralaz ine 2-8
80(吸
收)
0.3-0.7 0.5-1.5
1-4
(PO)
87 0.07
5-氟胞嘧
啶5-Flur ocytosin e 0.23 3-6
2-6
(PO)
48
氟哌啶醇
Haloperi dol 18-30 20-30
3-6
(PO)
0.72
氟氯青霉
素Fluclo xacillin 1.39 0.50.8 45
0.12-0.2
5
.04 96 0.2
氢化可的
松Hydroc
ortisone
0.462 0.5-2 100 .03 95 0.17 茶碱Theo0.115 3-9 >95 0.3-0.6 7.5-20 1-3 20 0.052
(PO) 保泰松Ph
enylbuta zone 24-120 90-100 0.1-0.35 40-150
1-2
(PO)
90-98 0.002
秋水仙碱
Colchici ne 0.462 0.5-1.5 2
0.5-2(P
O)
1.39
呋喃苯胺
酸(速尿)Furosemi de 1.4-3.5 0-60 0.1-0.2 0.1-0.2
1.0(PO)
0.5(IM)
95-99 0.042
奎尼丁Qu inidine 0.096 3-16 40-90 0.5-3.0 2-8
1-2
(PO)
30-90
min
(IM)
60-80 0.13
洋地黄毒
甙Digito xin 0.0058 96-168 90-100 0.5 0.01-0.035
1-3
(PO)
95-97 0.0026
美沙酮Me ygadone 0.091 7.6-15 100 1.5 0.48-0.86
4
(PO)
84-87 0.09
美多心安
Metoprol
ol
3-4 50 5.6 1.11 甲氨喋呤
Methotre xate 2-3.5
成人0.75
-0.8儿童
0.2-1.0
(PO)
1-3 50-70
4.3
5.5
氨喋青霉
素Ampici llin 0.6 1-1.5 50 0.4 2-8
0.5-1.0
(IM)
20 0.22
2.5(-COO
H)7.2(-N
H2)
氨酰心胺
Atenolol
6-9 50-60 0.7 0.2-1.3 2-4 0.065 胺甲苯辛0.35 2.5 30 3.2 2 0.89
lol
胍乙啶Gu
anethidi ne 43-120 50-80 3(OP)
8.3
11.4
胰岛素In sulin 10min 0.6
10-20μU/m
l
1-10 2.5
吲哚美辛
Indometh acin 2-10 9-100 0.3-1.6 0.5-3
1-2
(OP)
90 0.11
莨菪碱Hy
oscyamiu e 13-38 2-4
15-50min
(IM)1.0
(OP)
50 0.082 9.7
唑酮头孢
菌素Ceph
anone
0.277 2.5 0.47 88 0.047 萘啶酸Na
lidixic acid 0.43 1.5-2.0
0.26-0.4
5
93 0.14
羧苄青霉
素Carben icillin 0.462 1-1.5 <10 0.18 10-150
0.5-1.0
(IM)
50 0.071
2.7(-COO
H)2.7-3.
9(-NH2)
羟苄四唑
头孢菌素Cefamand ole 0.7 1.0 90-95 0.31
15min(I
V)0.5-1.
0(IM)
75 0.22
羟氨苄青
霉素Amox icillin 0.7 1.0-1.5 80-90 0.47 2-8 1.0(IM) 15-20 0.26
2.4(-COO
H)7.4(-N
H2)
硫酸奎宁
Quinine sulfate 0.13 5.3 100 2.0 2-5 1-2(PO) 70 0.26
4.1
8.4
紫霉素Vi
omycin
0.35 2 0.24 2(IM) 0.083 氯丙嗪
Chiorpro mazine 23-37 25 20 0.5-0.7
3-4(PO)
1-2(IM)
95-98 0.46 9.3
克林霉素
Clindamy cin 0.29 2-2.5 90 1.2 2-10
1.0(PO)
3.0(IM)
90 0.37
6.9
7.45
氯喹Chlo
roquine
0.013 70-120 80-100 112 6(PO) 55 0.82 8.4 氯硝基安
定Chlorn
itrazepa
m
20-60 75-90 1.5-4.4 1-2(PO) 45 0.052 10.8 氯霉素Ch
loramphe nicol 0.277 1.5-4 75-90
成人0.55
-0.98儿
童1.1-2.
5
4-20
2-3
(PO)
60-80 0.15 5.5
氯磺丙脲
Chlorpro
pamide
36-40 0.1-0.2 30-140 1-2(PO) 5-10 0.0027 5.0 普鲁卡因
胺Procai namide 2.5-4.5 75-95 1.7-2.2 4-8
1.0-2(P
O)30min
(IM)
15 0.4 9.2
卡巴咪嗪
Carbamaz epine(Te gretol) 20-60 0.8-1.4 2-10
8
(PO)
65-83 0.036
催产素Ox ytccin 5-16min 0.2
4-120(pg/m
l)
30 0.83
叠氮霉素
Azidocil
lin
0.866 0.6-1.0 0.35-0.6 83-85 0.43 2.8
美西律
(慢心
律)Mexil
etine
0.06 11.5 ≈100 4.5 0.5-20 0.27
新霉素Ne
omycin
0.35 2 0.25 1.0(IM) 0.087
溴苄胺Br
etylium
0.07 10 0.2-0.4
脱氧土霉
素Doxycy cline 0.03 14-25 90 1-2 1-10
1-3
(PO)
25-31 0.52 3.4
沷尼松Pr ednisone 0.693 1 0.5-0.6
1.0
(PO)
54 0.38
沷尼松龙
Predniso lone 0.217 3 0.5-0.6
1.0
(PO)
54 0.13
碳酸锂Li
thum car bonate 0.028 25 >95 0.7
0.4-0.7mmo
l/L
1-2
(PO)
0.02 6.8
链霉素St
reptomyc in 0.277 2-3 0.25
0.5-3(I
M)
30-35 0.07
镇痛新Pe
ntazocin e 0.375 1.85 3 0.03-0.1
1-3
(PO)
15-60
min
(IM)
60-70 1.12 11.2
磺胺Sulf anamide 0.077 9 80-85 100-150
2-4
(PO)
5-20 10.4
磺胺二甲胺基嘧啶
(SM2)Sulf amethoxa zole 0.099 7 0.61 80-100
2-4
(PO)
80 0.06 7.4
磺胺甲基异噁唑(S
MZ)Sulfa methoxaz ole 0.063 8-12 85-100 0.3 90-100
2-4
(PO)
68 0.021 6
磺胺异噁
唑(SIZ)S ulfameth oxazole 0.12 16 85-100 0.15 80-150
2-4
(PO)
68 0.017 6
磺胺嘧啶
0.041 17 85-100 0.6 2-4 68 0.025 6.3
(SD)
Sulfaiso
xazole
(PO)
噻吩甲氧
头孢菌素
Cefoxiti
n
0.99 0.7 90-95 0.29 0.005-0.01 同上45 0.29
噻吗心安
Timolol
2-4 72 1.8 1-2 73 0.42 噻嗪类利
尿药Thia zide 1-4 60-90
1-3
(PO)
99
简明常用血流动力学参数意义对照表 1. LSI 左心搏指数 2. RSI 右心搏指数 3. LCI 左心排指数 4. RCI 右心排指数 以上四个指数代表心脏的功能指数,其中左心排指数最重要,等同于心脏指数(CI),一般来说,CI<1.5=预后极差;1.5—2.0= 心源性休克;2.0—2.2=前向性心功能不全。 5. CWT 心脏总功率:反映心脏的负荷,一般运动时,功率会增大,如果正常情况总功率偏大,则代表心脏负荷偏大;偏小则视情况而定,有身体强健者,心脏功率不必很大,但器质性偏小,则有可能造成供血不足,头晕眼花等等。 6. LWE 左心室有效功率 7. LTPF 左心室总泵力 8. LWT 左心室功率 9. LEWK 左心室机械效率 10. JP 左心室喷血压力:该指数与血压有关,如果该指数偏大,则需要小心高血压了。 11. VP 左心室有效泵力 12. EF 喷血分数:非常重要的指标,EF值长期偏小,则有很大可能性是心衰。 13. AWK 动脉机械效率 14. EPE 射流压力 15. LCRI 左室等容指数 16. RCRI 右室等容指数 15/16两个参数代表心脏的容血量,其意义不如有效循环容量重要。 17. LVDV 左室舒张末血量 18. LVDP 左室舒末期压力 19. CR 左室喷血阻抗 20. PDM 平均舒张压:高血压的判断指标之一 21. PSM 平均收缩压:高血压的判断指标之一 22. PPM 平均脉压:高血压的判断指标之一 23. MAP 平均动脉压:高血压的判断指标之一 24. HR 心率 25. CVPS 中心静脉收缩压 26. CVPM 中心静脉平均压:非常重要的指标 严重升高:1.静脉充盈过量(循环超负荷) 2.静脉充血(心脏压塞、PEEP
药物代谢动力学(pharmacokinetics)简称药代动学或药动学,主要是定量研究药物在生物体内的过程(吸收、分布、代谢和排泄),并运用数学原理和方法阐述药物在机体内的动态规律的一门学科。确定药物的给药剂量和间隔时间的依据,是该药在它的作用部位能否达到安全有效的浓度。药物在作用部位的浓度受药物体内过程的影响而动态变化。在创新药物研制过程中,药物代谢动力学研究与药效学研究、毒理学研究处于同等重要的地位,已成为药物临床前研究和临床研究的重要组成部分。包括药物消除动力学:一级消除动力学(单位时间内消除的药量与血浆药物浓度成正比,又叫恒比消除)和零级消除动力学(单位时间内体内药物按照恒定的量消除,又叫恒量消除) 药物代谢动力学的重要参数: 1、药物清除半衰期(half life,t1/2),是血浆药物浓度下降一半所需要的时间。其长短可反映体内药物消除速度。 2、清除率(clearance,CL),是机体清除器官在单位时间内清除药物的血浆容积,即单位时间内有多少体积的血浆中所含药物被机体清除。使体内肝脏、肾脏和其他所有消除器官清除药物的总和。 3、表观分布容积(apparent volume of distribution,V d),是指当血浆和组织内药物分布达到平衡后,体内药物按此时的血浆药物浓度在体内分布时所需的体液容积。 4、生物利用度(bioavailability,F),即药物经血管外途径给药后吸收进入全身血液循环药物的相对量。可分为绝对生物利用度和相对生物利用度。 体内过程 即药物被吸收进入机体到最后被机体排出的全部历程,包括吸收、分布、代谢和排泄等过程。其中吸收、分布和排泄属物理变化称为转运。代谢属于化学变化亦称转化。机体对药物作用的过程,表现为体内药物浓度随时间变化的规律。药物动力学是研究药物体内过程规律,特别是研究血药浓度随时间而变化的规律。 1.吸收(absorption) 药物从给药部位进入血液循环的过程称为吸收。影响吸收的因素主要有: 1、给药途径:吸收速度:吸入>舌下>肌注>皮下>直肠>口服>皮肤。 2、药物性质: (1)脂溶性:脂溶性越大,吸收越快; (2)水溶性:易溶于水的药物易吸收; (3)离解度:不解离部分脂溶性较大,易吸收;而解离部分,由于带有极性,脂溶性低,难以吸收。。 口服药物被吸收进入体循环的比率,即给药量与吸收量的比率称为生物利用度(或生物可用度)。 2.分布(distribution) 药物吸收后从血液循环到达机体各个器官和组织的过程称为分布。 影响药物分布的主要因素有: 1、药物的性质:脂溶性大分布到组织器官的速度快。 2、药物与组织的亲和力:有些药物对某些组织器官有特殊的亲和力。药物对组织器官的亲和力与疗效及不良反应有关。
Sang-Wook Lee Biomedical Simulation Laboratory, University of Toronto, 5King’s College Road Toronto, Toronto,ON M5S3G8Canada; School of Mechanical and Automotive Engineering, University of Ulsan, Ulsan680-749,South Korea Luca Antiga Department of Bioengineering, Mario Negri Institute for Pharmacological Research, 24020Ranica(BG),Italy David A.Steinman1 Biomedical Simulation Laboratory, University of Toronto, 5King’s College Road Toronto, Toronto,ON M5S3G8Canada e-mail:steinman@mie.utoronto.ca Correlations Among Indicators of Disturbed Flow at the Normal Carotid Bifurcation A variety of hemodynamic wall parameters(HWP)has been proposed over the years to quantify hemodynamic disturbances as potential predictors or indicators of vascular wall dysfunction.The aim of this study was to determine whether some of these might,for practical purposes,be considered redundant.Image-based computational?uid dynamics simulations were carried out for N?50normal carotid bifurcations reconstructed from magnetic resonance imaging.Pairwise Spearman correlation analysis was performed for HWP quantifying wall shear stress magnitudes,spatial and temporal gradients,and harmonic contents.These were based on the spatial distributions of each HWP and, harmonic(DH)parameter were found to depend on how the wall shear stress magnitude was de?ned in the presence of?ow reversals.Many of the proposed HWP were found to provide essentially the same information about disturbed?ow at the normal carotid bifurcation.RRT is recommended as a robust single metric of low and oscillating shear. On the other hand,gradient-based HWP may be of limited utility in light of possible redundancies with other HWP,and practical challenges in their measurement.Further investigations are encouraged before these?ndings should be extrapolated to other vas-cular territories. ?DOI:10.1115/1.3127252? Keywords:wall shear stress,atherosclerosis,hemodynamic wall parameter,carotid bifurcation 1Introduction There is much evidence suggesting that initiation and progres-sion of atherosclerotic disease is in?uenced by“disturbed?ow”?1?.Notwithstanding the imprecise nature of this term?2?,various metrics have been proposed over the years to quantify?ow dis-turbances.Originally focused on the magnitudes of wall shear stress?WSS??3,4?these hemodynamic wall parameters?HWP?have since incorporated spatial and temporal gradients of WSS ?5–8?and,more recently,the harmonic content of time-varying WSS waveforms?2,9?. In a recent computational?uid dynamics?CFD?study of the relationship between geometry and disturbed?ow at the carotid bifurcations of young adults?10?,we noted that our?ndings were relatively insensitive to the choice of either time-averaged wall shear stress magnitude?TAWSS?or oscillatory shear index?OSI?as metrics of disturbed?ow.This was found to be explained by a strong and signi?cant inverse correlation between these two quan-tities.Such correlations among HWP are not unexpected,as rec-ognized early by Friedman and Deters?11?;however,they have been little-investigated in light of the growth in the number and complexity of candidate HWP. With this in mind,the objective of the present study was to use a representative sample of normal carotid bifurcation geometries to comprehensively test for correlations among established and recently-proposed HWP.Especially in the context of large-scale studies of so-called geometric and hemodynamic risk factors in atherosclerosis,we aimed to determine whether a subset of HWP, or even a single HWP,might serve as a suf?ciently robust marker of disturbed?ow. 2Materials and Methods 2.1Computational Fluid Dynamics.N=50anatomically re-alistic carotid bifurcation geometries were digitally reconstructed from black blood magnetic resonance imaging?MRI?of25osten- sibly healthy young adults,as described previously?12?.CFD simulations were carried out using a well-validated in-house ?nite-element-based CFD solver?13–15?.Quadratic tetrahedral- element meshes were generated by a commercial mesh generator ?ICEM-CFD;ANSYS,Berkeley,CA?using a nominally uniform node spacing of0.2mm,previously shown to be suf?cient for resolving wall shear stresses to within10%accuracy?16?.Rigid walls and Newtonian rheology were assumed.Pulsatile?ow boundary conditions were prescribed based on representative waveform shapes and allometrically-scaled inlet and outlet?ow rates.Further details of the CFD simulations are provided else- where?10?. For each tetrahedral element the vector WSS,?w,was calcu-lated as the projection of the stress tensor onto the element’s sur-face at each node,using the element’s quadratic shape functions. As nodes are connected to multiple elements,contributions to each nodal?w were averaged together.From these time-varying nodal WSS vectors,a variety of HWP were computed,as summa-rized in Table1,and detailed below. 1Corresponding author. Contributed by the Bioengineering Division of ASME for publication in the J OUR-NAL OF B IOMECHANICAL E NGINEERING.Manuscript received August12,2008;?nal manuscript received January1,2009;published online May11,2009.Review con-ducted by Fumihiko Kajiya.Paper presented at the2008Summer Bioengineering Conference?SBC2008?,Marco Island,FL,June25–29,2008.
一、吸收 溶出度:药物分子在消化道中溶解的程度 生物利用度:药物吸收的程度 绝对生物利用度 最大血药浓度(Cmax) 达峰时间(Tmax) 二、分布 由于体内环境的非均一性(血液、组织),导致药物浓度变化的速度不同。 隔室(compartment):同一隔室药物浓度的变化速度相同,均相。 一室模型:药物进入血液迅速分布全身,并不断被清除。 二室模型: 药物进入体内后,首先快速分布于组织中,然后进入较慢的消除过程。 表观分布体积(Vd)(aparent volume of distribution):表征药物在体内被组织摄取的能力。表观容积大的药物体内存留时间较长。 药物浓度-时间曲线下面积(AUC);系统药物暴露(Systemic Exposure) 血脑屏障;蛋白结合率;分布半衰期(t 1/2(α) 三、消除 消除(elimination):原药在体内消失的过程。包括肾(尿)或胆汁(粪)或呼吸排泄及代谢转化的总和。
消除速率常数(elimination constants):反映药物在体内消失的快慢。不完全反映药物的作用时间(代谢物也有活性)。 半寿期或半衰期(t1/2):药物浓度或药量降低50%所需的时间。消除半衰期t1/2(β))Terminal Half-life ,Elimination Half-life。 清除率(clearance,廓清率)或肾清除率(renal clearance):反映药物或代谢物经肾被排出体外的速度。 一方面是药物对机体的作用,产生药效、毒性或副作用,表现为药物的药理作用或毒理作用,决定于特定的化学结构,具有较强的结构特异性。 另一方面是机体对药物的作用:吸收、分布,生物转化和排泄,表现为药物的药代动力学性质。主要取决于药物的溶解性、脂水分配系数、电荷等药物分子整体的理化性质,结构特异性不强。 药物的吸收是药物由给药部位通过生物膜进入血液循环的过程。 吸收部位 消化道(口服给药,口腔、胃、小肠、大肠)、呼吸道(鼻腔给药,肺)、肌肉(肌肉注射)、粘膜(栓剂)。 吸收部位不同,药物被吸收的程度和快慢,有差异(静注、肌注;皮下给药,口服。) 共性:药物是通过生物膜吸收的。 吸收过程 扩散
药物代谢动力学完整版 第二章药物体内转运 肾脏排泄药物及其代谢物涉及三个过程:肾小球的滤过、肾小管主动分泌、肾小管重吸收。 一、药物跨膜转运的方式及特点 1. 被动扩散 特点:①顺浓度梯度转运②无选择性,与药物的油/水分配系数有关③无饱和现象④无竞争性抑制作用⑤不需要能量 2. 孔道转运 特点:①主要为水和电解质的转运②转运速率与所处组织及膜的性质有关 3. 特殊转运 包括:主动转运、载体转运、受体介导的转运 特点:①逆浓度梯度转运②常需要能量③有饱和现象④有竞争性抑制作用⑤有选择性 4. 其他转运方式 包括:①易化扩散类似于主动转运,但不需要能量②胞饮主要转运大分子化合物 二、影响药物吸收的因素有哪些 ①药物和剂型的影响②胃排空时间的影响③首过效应④肠上皮的外排⑤疾病⑥药物相互作用 三、研究药物吸收的方法有哪些,各有何特点? 1. 整体动物实验法 能够很好地反映给药后药物的吸收过程,是目前最常用的研究药物吸收的实验方法。缺点: ①不能从细胞或分子水平上研究药物的吸收机制; ②生物样本中的药物分析方法干扰较多,较难建立; ③由于试验个体间的差异,导致试验结果差异较大; ④整体动物或人体研究所需药量较大,周期较长。 2. 在体肠灌流法:本法能避免胃内容物和消化道固有生理活动对结果的影响。 3. 离体肠外翻法:该法可根据需要研究不同肠段的药物吸收或分泌特性及其影响因素。 4. Caco-2细胞模型法 Caco-2细胞的结构和生化作用都类似于人小肠上皮细胞,并且含有与刷状缘上皮细胞相关的酶系。优点: ①Caco-2细胞易于培养且生命力强,细胞培养条件相对容易控制,能够简便、快速地获得大量有价值的信息; ②Caco-2细胞来源是人结肠癌细胞,同源性好,可测定药物的细胞摄取及跨细胞膜转运; ③存在于正常小肠上皮中的各种转运体、代谢酶等在Caco-2细胞中大都也有相同的表达,因此更接近药物在人体内吸收的实际环境,可用于测定药物在细胞内的代谢和转运机制; ④可同时研究药物对粘膜的毒性; ⑤试验结果的重现性比在体法好。 缺点: ①酶和转运蛋白的表达不完整,此外来源,培养代数,培养时间对结果有影响; ②缺乏粘液层,需要时可与HT-29细胞共同培养。
药代动力学参数 This model paper was revised by the Standardization Office on December 10, 2020
一、吸收 溶出度:药物分子在消化道中溶解的程度 生物利用度:药物吸收的程度 绝对生物利用度 最大血药浓度(Cmax) 达峰时间(Tmax) 二、分布 由于体内环境的非均一性(血液、组织),导致药物浓度变化的速度不同。 隔室(compartment):同一隔室药物浓度的变化速度相同,均相。 一室模型:药物进入血液迅速分布全身,并不断被清除。 二室模型: 药物进入体内后,首先快速分布于组织中,然后进入较慢的消除过程。 表观分布体积(Vd)(aparent volume of distribution):表征药物在体内被组织摄取的能力。表观容积大的药物体内存留时间较长。 药物浓度-时间曲线下面积(AUC);系统药物暴露(Systemic Exposure) 血脑屏障;蛋白结合率;分布半衰期(t 1/2(α) 三、消除 消除(elimination):原药在体内消失的过程。包括肾(尿)或胆汁(粪)或呼吸排泄及代谢转化的总和。
消除速率常数(elimination constants):反映药物在体内消失的快慢。不完全反映药物的作用时间(代谢物也有活性)。 半寿期或半衰期(t1/2):药物浓度或药量降低50%所需的时间。消除半衰期t1/2(β))Terminal Half-life ,Elimination Half-life。 清除率(clearance,廓清率)或肾清除率(renal clearance):反映药物或代谢物经肾被排出体外的速度。 一方面是药物对机体的作用,产生药效、毒性或副作用,表现为药物的药理作用或毒理作用,决定于特定的化学结构,具有较强的结构特异性。 另一方面是机体对药物的作用:吸收、分布,生物转化和排泄,表现为药物的药代动力学性质。主要取决于药物的溶解性、脂水分配系数、电荷等药物分子整体的理化性质,结构特异性不强。 药物的吸收是药物由给药部位通过生物膜进入血液循环的过程。 吸收部位 消化道(口服给药,口腔、胃、小肠、大肠)、呼吸道(鼻腔给药,肺)、肌肉()、粘膜(栓剂)。 吸收部位不同,药物被吸收的程度和快慢,有差异(静注、肌注;皮下给药,口服。) 共性:药物是通过生物膜吸收的。 吸收过程 扩散
常用的药物代谢动力学参 数包括那些 Prepared on 24 November 2020
常用的药物代谢动力学参数包括那些. (1).表观分布容积 表示体内药量与血药浓度之间相互关系的一个比列常数。即体内药量按血浆中同样浓度分布时,所需体液的总容积。其数值反映了药物在体内的分布程度。表观分布容积是一个假设的容积,是假定药物在体内均匀分布情况下求得的药物分布容积,其意义在于:可计算出达到期望血浆药物浓度时的给药剂量;可以推测药物在体内的分布程度和组织中摄取程度。 (2).血浆药物浓度 指药物吸收后在血浆内的总浓度,包括与血浆蛋白结合的或在血浆游离的药物,有时也可泛指药物在全血中的浓度。药物作用的强度与药物在血浆中的浓度成正比,同时药物在血浆中的浓度也随时间变化。 (3).血药浓度—时间曲线 指给药后,以血浆(或尿液)药物浓度为纵坐标,时间为横坐标,绘制的曲线,简称药—时曲线,如图:
(4).血浆药物峰度浓度 简称峰浓度,指药—时曲线上的最高血浆药物浓度值,即用药后所能达到的最高血浆药物浓度,常以符号C max表示,单位以 ug/mL或者mg/L来表示。药物血浆浓度与药物的有效性与安全性直接相关。一般来说,峰浓度达到有效浓度才能显效,浓度越高效果越强,但超出安全范围则可出现毒性反应。另外,峰浓度还是衡量制剂吸收的一个重要指标。 (5).血浆药物浓度达峰时间 简称达峰时间,指在给药后人体血浆药物浓度曲线上达到最高浓度(峰浓度)所需时间,常以符号t max表示,单位一小时或分钟表示。达峰时间短,表示药物吸收快、起效迅速,但同时消除也快;而达峰时间长,则表示药物吸收和起效较慢,药物作用持续的时间也越长。达峰时间是应用药物和研究自己的一个重要指标。(6).血浆生物半衰期
药物代谢动力学 离子障 分子型的药物可自由穿透,而离子型药物被限制在生物膜的一侧,称为离子障 首过消除 药物通过胃肠壁和肝脏时可被代谢、失活,使进入体循环的药量减少,以口服途径给药最为常见 药物和血浆蛋白结合后对药物的影响 药理活性暂时消失,不能跨膜转运 药物与血浆结合特点 可逆性、非特异性、差异性、饱和性、竞争性 体内过程 药物的吸收、分布、代谢和排泄的总称,又叫药物的处置 消除 代谢和排泄的总称,是药物作用消失的主要原因 肝肠循环 有些药物在肝细胞内与葡萄糖醛酸等结合后通过胆汁排入小肠,在小肠被水解,部分药物可被再吸收重新进入血液循环的过程,称为肝肠循环。 肝药酶 存在肝细胞内质网中,促进药物转化的主要酶系统,主要是C色素P450 酶诱导剂(酶促剂) 能使肝药酶合成增加或活性增强的药物.如苯巴比妥、利福平
酶抑制剂(酶抑剂) 能使肝药酶合成减少或活性减弱的药物.如西咪替丁、异烟肼 酶促剂意义 使药物代谢加速,药效降低,常需增加剂量才能维持疗效。一旦停用药酶诱导剂,可是同服的药物浓度过高,药效增强,甚至中毒,是停药敏化现象的原因之一;还可加速自身代谢,是药物产生耐受性的原因之一;利用药酶诱导剂的酶促作用,可诱导新生儿肝药酶活性,促进血中游离胆红素与葡萄糖醛酸结合,经胆汁排出,用于预防新生儿脑核性黄疸 一级与零级消除动力学的差别: 一级消除动力学零级消除动力学 数学模式等比1000→500→250等差1000→900→800 发生条件给药量小于机体的消除能力给药量大于机体的消能力 半衰期恒定,与血药浓度无关不恒定,与血药浓度有关 时量曲线曲线直线 直线曲线 对数时量曲 线 量效关系增加药物的剂量增加药物的剂量 药物作用时间呈低比例延长药物作用时间呈超比例延长 曲线下面积AUC 药物时-量曲线下的面积,AUC大小与进入体循环的药量成正比,反应进入体循环药物的相对量 半衰期 指血浆药物浓度下降一半所需时间 意义 确定给药时间;估计达到稳态血药浓度Css所需时间;估计停药后药物体内消除所需时间;按半衰期时间的长短对药物分类;反映药物消除快慢程度。 表观分布容积
眼部药物代谢动力学的研究 摘要:药代动力学研究在药品研发及评价过程中具有重要作用。本文对眼用制剂的药代动力学研究的进行了探讨。本文介绍了眼的药物代谢途径及其特点,眼部药代动力学样本采集以及常见的分析方法。通过药代动力学的研究可以了解药物在体内的代谢特征,指导临床用药的给药剂量、间隔、途径等,保证药物疗效,减少毒副作用。 关键词:眼;药代动力学;分析方法 对药品研发人员、技术审评人员以及临床医师来说, 人体药代动力学研究所提供的信息是非常重要的, 有时甚至是其他研究所无法替代的。通过眼部药代动力学的研究,学者们可以获得药物在眼部代谢的浓度一时间曲线,了解药物在眼内的代谢特征,对于药物剂型设计、给药方案、治疗监测等起着重要的作用。但是眼具有特殊屏障作用, 眼用制剂的药代动力学过程不同于系统给药的过程, 那么其药代动力学的特点及一般过程如何? 其意义何在? 常用的分析方法用哪些?另外,现有眼科局部给药的突破点在于,如何通过改进药物剂型和给药方式保证病人良好的依从性和药物应用的便捷性, 安全性。因此,对眼局部的药物代谢动力学加以了解和考察,能够更好的针对不同的疾病选择不同的给药方式和药物剂型, 对于临床诊治及用药安全具有重要的指导意义。同时,眼部给药系统研究的兴起也要求有方便、准确、安全、有效的检测方法来进行说明和验证。 1 眼的药物代谢途径及其特点 1.1 药物在眼球表面的流失 在眼局部应用滴眼剂时, 由于泪液在眼表的涂布,流动和循环使药物在接触眼表时即发生了流失。泪液在眼表的更新速度仅为1μl/min,相对多量的滴眼剂可在数分钟之内随着泪液的循环快速流入鼻泪管发生排泄[1]。由此可见,滴眼剂药物的全身吸收不仅在结膜囊的局部毛细血管中进行,同时也存在于滴眼剂流入鼻腔从而进入全身的情况。因此, 大多数以局部滴眼剂方式给药的小分子量药物在数分钟内即会进入全身循环,从而导致其在眼表的生物利用度仅在5%甚至更少。全身循环中的吸收降低了药物在眼表的有效浓集, 因此,通过应用固体药物
药物代动力学完整版 第二章药物体转运 肾脏排泄药物及其代物涉及三个过程:肾小球的滤过、肾小管主动分泌、肾小管重吸收。 一、药物跨膜转运的方式及特点 1. 被动扩散 特点:①顺浓度梯度转运②无选择性,与药物的油/水分配系数有关③无饱和现象④无竞争性抑制作用⑤不需要能量 2. 孔道转运 特点:①主要为水和电解质的转运②转运速率与所处组织及膜的性质有关 3. 特殊转运 包括:主动转运、载体转运、受体介导的转运 特点:①逆浓度梯度转运②常需要能量③有饱和现象④有竞争性抑制作用⑤有选择性 4. 其他转运方式 包括:①易化扩散类似于主动转运,但不需要能量②胞饮主要转运大分子化合物 二、影响药物吸收的因素有哪些 ①药物和剂型的影响②胃排空时间的影响③首过效应④肠上皮的外排⑤疾病⑥药物相互作用 三、研究药物吸收的方法有哪些,各有何特点? 1. 整体动物实验法 能够很好地反映给药后药物的吸收过程,是目前最常用的研究药物吸收的实验方法。缺点: ①不能从细胞或分子水平上研究药物的吸收机制; ②生物样本中的药物分析方法干扰较多,较难建立; ③由于试验个体间的差异,导致试验结果差异较大; ④整体动物或人体研究所需药量较大,周期较长。 2. 在体肠灌流法:本法能避免胃容物和消化道固有生理活动对结果的影响。 3. 离体肠外翻法:该法可根据需要研究不同肠段的药物吸收或分泌特性及其影响因素。 4. Caco-2细胞模型法 Caco-2细胞的结构和生化作用都类似于人小肠上皮细胞,并且含有与刷状缘上皮细胞相关的酶系。优点: ①Caco-2细胞易于培养且生命力强,细胞培养条件相对容易控制,能够简便、快速地获得大量有价值的信息; ②Caco-2细胞来源是人结肠癌细胞,同源性好,可测定药物的细胞摄取及跨细胞膜转运; ③存在于正常小肠上皮中的各种转运体、代酶等在Caco-2细胞都也有相同的表达,因此更接近药物在人体吸收的实际环境,可用于测定药物在细胞的代和转运机制; ④可同时研究药物对粘膜的毒性; ⑤试验结果的重现性比在体法好。 缺点: ①酶和转运蛋白的表达不完整,此外来源,培养代数,培养时间对结果有影响; ②缺乏粘液层,需要时可与HT-29细胞共同培养。
常用的药物代谢动力学 参数包括那些 Document number【980KGB-6898YT-769T8CB-246UT-18GG08】
常用的药物代谢动力学参数包括那些. (1).表观分布容积 表示体内药量与血药浓度之间相互关系的一个比列常数。即体内药量按血浆中同样浓度分布时,所需体液的总容积。其数值反映了药物在体内的分布程度。表观分布容积是一个假设的容积,是假定药物在体内均匀分布情况下求得的药物分布容积,其意义在于:可计算出达到期望血浆药物浓度时的给药剂量;可以推测药物在体内的分布程度和组织中摄取程度。 (2).血浆药物浓度 指药物吸收后在血浆内的总浓度,包括与血浆蛋白结合的或在血浆游离的药物,有时也可泛指药物在全血中的浓度。药物作用的强度与药物在血浆中的浓度成正比,同时药物在血浆中的浓度也随时间变化。 (3).血药浓度—时间曲线 指给药后,以血浆(或尿液)药物浓度为纵坐标,时间为横坐标,绘制的曲线,简称药—时曲线,如图:
(4).血浆药物峰度浓度 简称峰浓度,指药—时曲线上的最高血浆药物浓度值,即用药后所能达到的最高血浆药物浓度,常以符号Cmax表示,单位以ug/mL或者mg/L来表示。药物血浆浓度与药物的有效性与安全性直接相关。一般来说,峰浓度达到有效浓度才能显效,浓度越高效果越强,但超出安全范围则可出现毒性反应。另外,峰浓度还是衡量制剂吸收的一个重要指标。 (5).血浆药物浓度达峰时间 简称达峰时间,指在给药后人体血浆药物浓度曲线上达到最高浓度(峰浓度)所需时间,常以符号tmax表示,单位一小时或分钟表示。达峰时间短,表示药物吸收快、起效迅速,但同时消除也快;而达峰时间长,则表示药物吸收和起效较慢,药物作用持续的时间也越长。达峰时间是应用药物和研究自己的一个重要指标。 (6).血浆生物半衰期
血流动力学参数 关键词:血流动力学监测,参数,运用,意义 摘要:血流动力学监测已经广泛应用于危重症,但其数据多,每个参数因为它来源的监测手段和影响因素等等的不同,其临床意义不同。血流动力学监测的每个参数都有他的“背景”、有它的长处和短处。哪个参数有什么意义,怎样结合其它参数或临床等等都是我们应该掌握和经常思考的,而且只有在临床中不断运用、思考才能真正理解这些参数。本文介绍了直接测量所得指标:上肢动脉血压、心率、中心静脉压、右心房压、右心室压、肺动脉压、肺毛细血管嵌顿压、心输出量。由直接测量指标所派生的指标:心脏排血指数、心脏搏出量、肺血管阻力、心室做功指数和PICCO参数:血管外肺水、胸内血容量。介绍了临床应用于判断左心功能、疾病的鉴别、心功能状态的治疗原则、指导疾病的治疗等。 1、主要监测指标 1.1直接测量所得指标 1.1.1上肢动脉血压(AP) 正常值:收缩压1 2.0~18.7kPa(90~140mmHg),舒张压8.0~12.0kPa(60~90mmHg)。心排量、全身血管阻力、大动脉壁弹性、循环容量及血液粘度等均可影响动脉血压。一般用袖带血压计测量。在休克或体循环直视心脏手术时,应以桡动脉穿刺直接测量为准[1]。血压是反应心排量水平和保证器官有效灌注的基础,过高时增大左室后负荷和心肌耗氧,过低不能保证重要器官有效灌注。当MAP低于75mmHg 时,心肌供血曲线变陡下降,因此,MAP75~80mmHg,是保证心肌供血大致正常的最低限度[2]。对原有高血压病人,合理的MAP应略高于此。 1.1.2心率(HR)正常值:60~100次/min。反映心泵对代谢改变、应激反应、容量改
药代主要参数标准化工作室编码[XX968T-XX89628-XJ668-XT689N]
药物代谢动力学的主要参数 根据时间-药物浓度曲线,采用相应的药代动力学计算机程序包进行数学处理,可估算 出药物在体内吸收、分布、转化和排泄等相关的若干药代动力学参数(pharmacokineticparameters),以反映药物在体内的动力学规律和特点。常用的药代动力学参数有: (一)药峰时间(Tmax)和药峰浓度(Cmax) 药峰时间(Tmax)是指用药以后,血药浓度达到峰值所需的时间。药峰时间短,表示药物吸收快、起效迅速,但同时消除也快;药峰时间长,则表明药物吸收和起效较慢,但作用持续时间也往往较长(图3-7)。药峰时间是研究药物制剂的一个重要指标。 药峰浓度(Cmax)又称峰值(peakvalue),是指用药后所能达到的最高血药浓度(见图3-9)。药峰浓度与药物的临床应用密切相关,药峰浓度要达到有效浓度才能显效,但若高出安全范围则可表现为毒性反应。 (二)时量曲线下面积(areaunderthetimeconcentrationcurve,AUC) 又称曲线下面积,是指由坐标横轴与时间-药物浓度曲线围成的面积。它代表一段时间 内,血液中的药物的相对累积量,也是研究药物制剂的一个重要指标(图3-11)。其单位为μg/ml·h,通常采用梯形法计算,计算公式为: (三)生物利用度(bioavailability,afractionofdose,F) 生物利用度是指血管外给药时,药物吸收进入血液循环的相对数量。生物利用度也是 评价药物制剂质量的一个十分重要的指标。通常用吸收百分率表示,即给药量与吸收进入体循环的药量的比值(见式①): 相对F是评价厂家产品质量的重要标准之一。如果制剂质量不合格,生物利用度低, 临床疗效肯定差。一般药典上都规定药厂生产的制剂,生物利用度的差距不应超过 ±10%。生物利用度的意义在于:①从制剂方面而言,剂量和剂型相同的药物,如果厂家的制剂工艺不同,甚至同一药厂生产的同一制剂的药物,仅因批号不同,都可以使药物的晶型、颗粒大小或其他物理特性,以及药物的生产质量控制等发生改变,从而影响药物的崩解和溶解度,使药物的生物利用度发生明显的改变,导致时间-药物浓度曲线的改变(见图3-12)。②从机体方面而言,剂量、剂型甚至制剂都完全相同的药物,因为在不同生理或病理条件下应用,也可引起生物利用度的改变,使时间-药物浓
1 药物代谢动力学 一、药物代谢动力学 简称药动学或药代学,研究机体对药物的作用,药物在体内吸收、分布、代谢(生物转化)和排泄及血浆药物浓度动态变化规律的科学。 ● 药物转运:药物吸收、分布和排泄,仅是药物在体内位置的迁移。 ● 药物转化:即药物代谢,是药物在体内发生化学结构的变化。 一、药物的体内过程 1.药物吸收及影响的因素 吸收概念:是指药物从给药部位进入血液循环的过程— 静脉给药没有吸收过程 吸收途径:血管外给药途径有——消化道给药、注射给药、呼吸道给药和皮肤黏膜给药。 (1)消化道吸收: 包括口腔吸收、胃吸收、小肠吸收、直肠吸收。其中小肠是药物吸收的主要部位(原因有 3:吸收面积大、血流丰富、pH 适当,既适合酸性药物的吸收,又适合碱性药物的吸收。) (2)注射部位的吸收: ① 肌内注射 ② 皮下注射—不包括静脉注射 ③ 其他注射给药:包括动脉注射和鞘内注射(将药物注射到脊髓的蛛网膜下腔,如进行脊髓麻醉) (3)肺部吸收:挥发性或气体性药物通过肺上皮细胞或气管黏膜吸收。 (4)经皮吸收: 局部给药, 吸收的速率和程度取决于用药的面积、药物的脂溶性及皮肤受损情况。 【影响吸收的因素】 (1)药物的理化性质:弱酸性药物在胃中易吸收,而弱碱性药物在小肠中吸收。 药物吸收与排泄的规律是酸酸碱碱促吸收,酸碱碱酸促排泄。 (2)药物的剂型 药物制剂释放速率和溶解速率影响药物的吸收。 (3)首过(关)消除:某些药物在通过胃肠黏膜及肝脏时,部分被代谢失活,进入体循环的药量减少,称为首过消除或首关效应。 ● 如硝酸甘油、利多卡因、异丙肾上腺素都具有明显的首过消除。 ● 掌握受过消除的意义:首关消除明显的药物不能采取口服给药。 (4)吸收环境 主要涉及胃肠内容物、胃肠液酸碱度、胃肠蠕动和排空、血流量等。 2.药物分布及其影响因素 (1)概念:药物进入血液后,随血液运至机体各组织的过程。 (2)影响因素 ① 药物与血浆蛋白结合:药物进入血液后,与血浆蛋白结合成为结合型药物, 未被结合的药物则称为游离型药物。血浆蛋白结合率:即血中与蛋白结合 的药物占总药量的百分数,表示药物与血浆蛋白结合的程度。 ② 机体特殊屏障 1)血脑屏障 血脑屏障是血液-脑细胞、血液-脑脊液及脑脊液-脑细胞三种屏障的总称。能阻碍药物穿透的主要是前二者。血脑屏障的物质基础——与其他组织器官的毛细血管相比,脑毛细血管内皮细胞紧密相连,内皮细胞之间无间隙,彼此重叠覆盖,连接紧密。表面又被星形胶质细胞所包围,比一般的毛细血管壁多了一层胶质细胞。血脑屏障是保护大脑的生理屏障。 只有分子量较小、脂溶性较高的药物才有可能通过血脑屏障而进入脑组织。
第二章药物代谢动力学 一、A 1、合理用药需要具备下述药理学知识 A.药物的作用与副作用 B.药物的毒性与安全范围 C.药物的效价与效能 D.药物的半衰期与消除途径 E.以上均需要 2、决定药物每天用药次数的主要因素是 A.吸收快慢 B.作用强弱 C.体内分布速度 D.体内转化速度 E.体内消除速度 3、药时曲线下面积代表 A.药物血浆半衰期 B.药物的分布容积 C.药物吸收速度 D.药物排泄量 E.生物利用度 4、需要维持药物有效血浓度时,正确的恒定给药间隔时间是 A.每4h给药一次 B.每6h给药一次 C.每8h给药一次 D.每12h给药一次 E.每隔一个半衰期给药一次 5、下述叙述中错误的是 A.在给药方法中口服最常用 B.过敏反应难以预知 C.药物的作用性质与给药途径无关 D.肌注比皮下注射快 E.大多数药物不易经皮肤吸收 6、以近似血浆半衰期的时间间隔给药,为迅速达到稳态血浓度,可以首次剂量 A.增加半倍 B.增加1倍 C.增加2倍 D.增加3倍 E.增加4倍
7、下列关于恒量、定时分次注射给药时,血浆药物浓度变化的描述中错误的是 A.要5个半衰期达到Css B.峰值((Cmax)与谷值(Cmin)波动之比与间隔时间(t)有关 C.t越短波动越小 D.药量不变,t越小可使达到Css时间越短 E.肌注时Cmax与Cmin的波动比静注时大 8、恒速静脉滴注按一级动力学消除的药物时,达到稳态浓度的时间取决于 A.静滴速度 B.溶液浓度 C.血浆半衰期 D.药物体内分布 E.血浆蛋白结合量 9、某药的半衰期是7h,如果按每次0.3g,一天给药3次,达到稳态血药浓度所需时间是 A.5~10h B.10~16h C.17~23h D.24~28h E.28~36h 10、按一级动力学消除的药物,按一定时间间隔连续给予一定剂量,达到稳态血药浓度时间长短决定于 A.剂量大小 B.给药次数 C.吸收速率常数 D.表观分布容积 E.消除速率常数 11、某药半衰期为8h,一日3次给药,达到稳态血药浓度的时间为 A.0.5~1d B.1.5~2.5d C.2.5~3.5d D.3.5~4.5d E.4.5~5.5d 12、恒量恒速给药最后形成的血药浓度为 A.有效血浓度 B.稳态血药浓度 C.峰浓度 D.阈浓度 E.中毒浓度