外消旋氯吡格雷的合成
- 格式:pdf
- 大小:157.56 KB
- 文档页数:2

氯吡格雷外消旋体合成的研究进展前言随着生活水平的提高和生活方式的改变,心脑血管疾病已成为人类健康的主要疾病,在我国,尤其是脑血管疾病,患病率高达490人/10万人[1]。
研究表明,此类疾病的基础病因是动脉粥样硬化,而血小板抑制剂能有效地对抗动脉粥样硬化血栓栓塞性疾病。
虽然阿司匹林和噻氯匹定(抵克立得)在抑制血栓形成方面均有疗效,但二者都具有潜在的严重不良反应。
在此基础上研发了新一代血小板聚集抑制剂—氯吡格雷。
药用其硫酸氢盐。
氯吡格雷是噻吩吡啶的衍生物,具有对骨髓抑制作用少等优点。
该产品于1998年3月率先在美国上市,随后进入欧洲、北美、澳洲、新加坡等多国市场,并于2001年8月在中国上市[2]。
作为新一代的血小板聚集抑制剂,氯吡格雷具有以下两个特点:(1)药理特点氯吡格雷属于噻吩吡啶类衍生物,通过选择性的与血小板表面腺苷酸环化酶偶联的二磷酸腺苷(ADP)受体结合,抑制纤维蛋白原与血小板受体GP Ⅱb/Ⅲa结合而发挥作用[3]。
药理研究表明,氯吡格雷具有较噻氯匹定和阿司匹林更优异的药理活性,其活性强而持久。
因其很强的抗ADP诱导的血小板聚集作用及对静、动脉血栓形成的抑制,使它能治疗和预防动脉粥样硬化、动静脉狭窄、损伤、手术等各种因素引起的血小板聚集性血栓,预防和治疗中风、心梗等心脑血管及外周动脉血管疾病。
有更好的应用前景[4]。
(2)安全特点全球范围内的大量大规模的临床研究证实了氯吡格雷的疗效及安全性。
其中,成功的大型临床氯吡格雷应用于缺血性疾病高危患者的比较研究是氯吡格雷取得快速批准的关键。
研究收入了200例新近患心肌梗死脑卒中或已确诊的外周动脉疾病由于下肢供血不足引起疼痛患者,以评价氯吡格雷的疗效及安全性。
最终结果表明氯吡格雷在降低缺血性脑卒中、心肌梗死、心血管性死亡的相对危险性较阿司匹林优,尤其在降低心肌梗死的相对危险性方面,较阿司匹林优。
在安全性方面,使用氯吡格雷的总体安全性情况良好,较阿司匹林具有更好的消化道安全性和耐受性中性粒细胞减少的发生率在阿司匹林组与氯吡格雷组中相似,不良反应较少较轻,主要表现为上腹不适,偶见皮疹、皮肤粘膜出血,罕见白细胞减少和粒细胞缺乏。


氯吡格雷合成工艺氯吡格雷合成工艺介绍•氯吡格雷是一种血小板凝集抑制剂,可用于预防和治疗血栓性疾病。
•氯吡格雷的合成工艺是制备氯吡格雷的重要步骤。
步骤1.原材料准备–2-甲氧基苯基乙酸–氯乙酸–氯丙酸–辛醇–硫酸–氯丁酸–氯化亚砜–叠氮化钠–硝酸2.第一步:合成2-甲氧基苯基乙酸氯乙酯–将2-甲氧基苯基乙酸、氯乙酸、辛醇等原材料混合反应,生成2-甲氧基苯基乙酸氯乙酯。
3.第二步:合成2-甲氧基苯基乙酸氯吡格雷–将第一步得到的2-甲氧基苯基乙酸氯乙酯与氯丙酸、硫酸等原材料混合反应,生成2-甲氧基苯基乙酸氯吡格雷。
4.第三步:合成氯吡格雷–将第二步得到的2-甲氧基苯基乙酸氯吡格雷与氯丁酸、氯化亚砜等原材料混合反应,生成氯吡格雷。
5.第四步:纯化与结晶–对反应得到的氯吡格雷进行纯化与结晶,以提高纯度和净化产物。
结论•氯吡格雷合成工艺经过多个步骤的反应和处理,最终得到高纯度的氯吡格雷。
•该合成工艺可为氯吡格雷的生产提供可靠的方法和技术支持,为临床应用提供保障。
请注意:以上仅为示例内容,请在实际撰写文章时根据相关资料进行适当的修改和补充。
优点•合成工艺相对较简单,原材料易得并且成本较低。
•通过纯化与结晶可提高产品纯度,提供高质量的氯吡格雷。
•生产过程中有较少的副产物生成,减少环境污染。
•可大规模生产,满足市场需求。
挑战与改进•在合成过程中,有可能出现杂质产生或无法完全转化的情况,需要进一步优化反应条件和操作步骤。
•提高反应收率和选择性,减少催化剂的使用量,降低生产成本。
•进一步优化纯化与结晶过程,提高产物收率和纯度。
•加强工艺控制,确保生产安全和产品质量稳定性。
应用前景•氯吡格雷是一种广泛应用于临床的抗血小板药物。
•随着人们对心血管疾病的认识不断深化,对抗血小板药物的需求也在增加。
•氯吡格雷具有较少的副作用和良好的效果,作为一种高效、安全的药物,其应用前景广阔。
总的来说,氯吡格雷合成工艺的研究和发展对于药物的生产和应用具有重要意义,可以提供高质量的氯吡格雷,满足临床需求。

氯吡格雷的生产工艺与技术路线的选择2.1 氯吡格雷主要生产方法2.1.1先合成后拆分法在1985年Sanofi公司发表的专利上,氯吡格雷的早期合成方法,是用4, 5, 6, 7-四氢噻吩并[3, 2-c]吡啶与α-氯(2-氯)苯乙酸甲酯在碳酸钾和四氢呋喃存在下反应生成氯吡格雷的外消旋体,然后进行手性拆分,该路线中合成消旋氯吡格雷的收率仅有45%。
随后1991年Sanofi公司对上述路线进行了改进,用α-溴(2-氯)苯乙酸甲酯代替α-氯(2-氯)苯乙酸甲酯进行反应,产率有较大提高。
该路线首先分别合成出4, 5, 6, 7-四氢噻吩并[3, 2-c]吡啶和α-溴(2-氯)苯乙酸甲酯,二者反应生成氯吡格雷的外消旋体,然后进行手性拆分(式1)。
该法中用到的两个反应物的生产工艺都比较成熟,其中4, 5, 6, 7-四氢噻吩并[3, 2-c]吡啶同时也是生产噻氯匹定的重要中间体。
该法是氯吡格雷的早期合成方法,虽然其工艺比较简单,原料价廉易得,比较适合于生产噻氯匹定的药厂,但收率还不理想;另外放在最后进行手性拆分,目标产物的收率最多仅为产物的一半。
2.1.2先缩合再环合法…此处省略,详情请见六鉴网()《氯吡格雷技术与市场调研报告》2.1.3先拆分后合成法该方法以邻氯扁桃酸为原料,首先进行拆分,甲酯化后与苯磺酰氯反应生成具有强的离去基团(-OSO2Ph)的手性中间体,然后与4, 5, 6, 7-四氢噻吩并[3, 2-c]吡啶在碳酸钾的催化下,发生双分子亲核取代反应,构型翻转生成氯吡格雷(式7)。
专利报道,该路线中甲酯化反应、生成磺酸酯的反应和最后一步的亲核取代反应,每步的收率均在90%以上;且前两步反应基本无消旋化,最后一步的光学纯度也超过90%。
综上所述,在上述几种氯吡格雷的合成方法中,以先拆分后合成法效果最佳。
所用的起始原料邻氯扁桃酸易得,后续的几步都是比较成熟的反应,操作简便且收率较高,该法最大的优势是把手性拆分放在反应前进行,大大提高了最终产物的收率。
![氯吡格雷新生产工艺[发明专利]](https://uimg.taocdn.com/97831ed83968011ca2009199.webp)
专利名称:氯吡格雷新生产工艺专利类型:发明专利
发明人:叶红平,朱作霖,孙盟
申请号:CN200510053258.9申请日:20050218
公开号:CN1778936A
公开日:
20060531
专利内容由知识产权出版社提供
摘要:本发明公开了一种氯吡格雷新生产工艺,即生化合成工艺,解决了现有合成方法要进行重结晶拆分才能得到需要(S)-对映体的问题。
本发明利用氰化物水解酶催化水解外消旋的有机氰化物成相应的具有高旋光度的有机酸,再经过系列反应直接合成(S)-对映体氯吡格雷,其工艺过程中不用拆分,因而收率高,成本低,简化了工艺。
申请人:淮北市辉克药业有限公司
地址:235000 安徽省淮北市惠黎路惠黎工商大厦六楼
国籍:CN
更多信息请下载全文后查看。


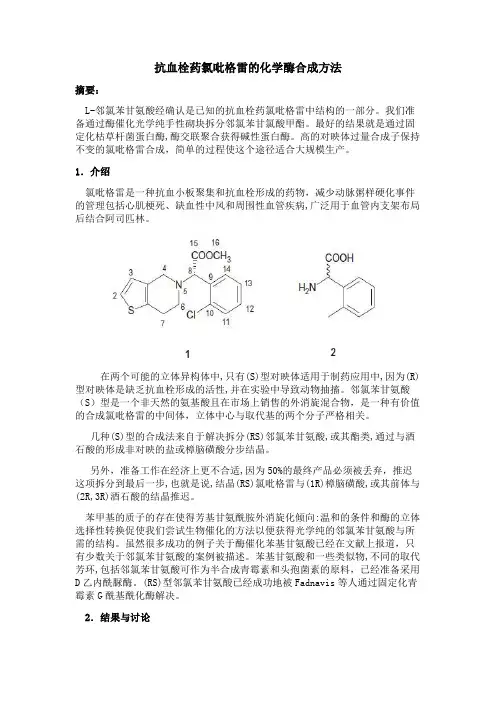
抗血栓药氯吡格雷的化学酶合成方法摘要:L-邻氯苯甘氨酸经确认是已知的抗血栓药氯吡格雷中结构的一部分。
我们准备通过酶催化光学纯手性砌块拆分邻氯苯甘氯酸甲酯。
最好的结果就是通过固定化枯草杆菌蛋白酶,酶交联聚合获得碱性蛋白酶。
高的对映体过量合成子保持不变的氯吡格雷合成,简单的过程使这个途径适合大规模生产。
1.介绍氯吡格雷是一种抗血小板聚集和抗血栓形成的药物,减少动脉粥样硬化事件的管理包括心肌梗死、缺血性中风和周围性血管疾病,广泛用于血管内支架布局后结合阿司匹林。
在两个可能的立体异构体中,只有(S)型对映体适用于制药应用中,因为(R)型对映体是缺乏抗血栓形成的活性,并在实验中导致动物抽搐。
邻氯苯甘氨酸(S)型是一个非天然的氨基酸且在市场上销售的外消旋混合物,是一种有价值的合成氯吡格雷的中间体,立体中心与取代基的两个分子严格相关。
几种(S)型的合成法来自于解决拆分(RS)邻氯苯甘氨酸,或其酯类,通过与酒石酸的形成非对映的盐或樟脑磺酸分步结晶。
另外,准备工作在经济上更不合适,因为50%的最终产品必须被丢弃,推迟这项拆分到最后一步,也就是说,结晶(RS)氯吡格雷与(1R)樟脑磺酸,或其前体与(2R,3R)酒石酸的结晶推迟。
苯甲基的质子的存在使得芳基甘氨酰胺外消旋化倾向:温和的条件和酶的立体选择性转换促使我们尝试生物催化的方法以便获得光学纯的邻氯苯甘氨酸与所需的结构。
虽然很多成功的例子关于酶催化苯基甘氨酸已经在文献上报道,只有少数关于邻氯苯甘氨酸的案例被描述。
苯基甘氨酸和一些类似物,不同的取代芳环,包括邻氯苯甘氨酸可作为半合成青霉素和头孢菌素的原料,已经准备采用D乙内酰脲酶。
(RS)型邻氯苯甘氨酸已经成功地被Fadnavis等人通过固定化青霉素G酰基酰化酶解决。
2.结果与讨论2.1酶催化RS型邻氯苯甘氨酸的拆分首先,我们计划去探讨生物阴极转换羧基或氨基的可能性,即酶催化水解或合成酯3和酰胺4。
表1酶法水解(RS)31.高效液相色谱法计算。

氯吡格雷合成工艺(一)氯吡格雷合成工艺核心原料•卡格雷(CAS号:•氯乙醇(CAS号:•氨(CAS号:•溴化亚铁(CAS号:合成步骤1.预处理•将卡格雷溶于氯乙醇中,搅拌均匀,得到溶液A。
•在溴化亚铁中加入适量的氨溶液,搅拌至溴化亚铁完全溶解,得到溶液B。
2.反应•将溶液A缓慢加入溶液B中,同时控制反应温度在40-50摄氏度,搅拌反应3小时。
•反应结束后,将反应液经过冷却、过滤、洗涤等处理,得到初步产品。
3.纯化•初步产品经过晶体分离、溶剂结晶、洗涤、干燥等工艺流程,纯化得到氯吡格雷产物。
•最终产物经过粉碎、包装等过程,得到合成的氯吡格雷粉末。
工艺优势•简单:合成步骤较少,主要通过溶液反应得到产品。
•高产率:通过优化反应条件和工艺流程,可获得高纯度、高产率的氯吡格雷产物。
•可扩展性:工艺可根据生产需求进行调整和优化,适合大规模合成。
实际应用•氯吡格雷,作为一种抗凝血药物,广泛应用于心脑血管疾病的预防和治疗。
•由于其药效明显且不良反应较少,氯吡格雷在医药领域受到广泛重视和应用。
•合成工艺的优化,不仅提高了氯吡格雷的可获得性,也为其规模化生产提供了便利。
总结•氯吡格雷合成工艺通过简单的溶液反应和纯化工艺,可实现高效、高产的合成。
•优化工艺流程和反应条件,提高了合成产物的纯度和产率。
•氯吡格雷的合成工艺为其广泛应用于心脑血管疾病的治疗提供了可靠的来源。
工艺优化•反应温度优化:通过调节反应温度,可控制反应速率和产物纯度,进一步提高工艺效率。
•溶剂选择优化:选用适合的溶剂可以提高反应物的溶解度和反应效率。
•搅拌条件优化:合适的搅拌条件可保证反应物均匀混合,利于反应进行。
•结晶技术优化:采用合适的结晶技术,如控制结晶温度和时间,可提高产物的晶体纯度。
未来发展方向•绿色化生产:推动氯吡格雷合成工艺的绿色化,减少对环境的影响,提高可持续性。
•新型催化剂研究:探索新型催化剂的应用,提高反应速率和产物纯度。
•自动化生产:引入自动化技术,提高生产效率和稳定性,降低人为误差。


Vol.41No.5(2010)
ZHEJIANG CHEMICAL INDUSTRY 外消旋氯吡格雷的合成
许珂慧刘蓓蓓林斐斐潘冰凡郭海昌(台州学院医药化工学院,浙江
临海
317000)
摘要:以噻吩乙胺为起始原料,经Pictet Spengler 反应得4,5,6,7-四氢噻吩并[3,2-C]吡啶盐酸盐,
然后和α-溴代邻氯苯乙酸甲酯反应得外消旋氯吡格雷,总收率达61.5%。
关键词:氯吡格雷;2-噻吩乙胺;Pictet Spengler ;α-溴代邻氯苯乙酸甲酯;合成
收稿日期:2010-02-04
作者简介:郭海昌(1976-),男,讲师,从事药物及中间体的合成研究。
文章编号:1006-4184(2010)05-0006-02
农药·医药化工
氯吡格雷是高效的抗血小板凝聚剂,临床用于预防心肌梗塞、中风及动脉粥样硬化,作用强度高,副作用低[1-3]。
该药在欧美发达国家己大量使用,但由于价格昂贵,在我国还处在推广阶段。
文献报道的氯吡格雷合成路线较多,按其起始原料或关键中间体的不同可分成两大类:一类是以噻吩乙醇为起始原料,主要和邻氯苯甘氨酸等经几
步反应后得到产物[4,5];另一类以噻吩乙胺为起始原料,主要和邻氯扁桃酸或α-卤代邻氯苯乙酸等经几步反应后得到产物[6-10]。
本文采用后者路线,以噻吩乙胺为起始原料,经Pictet Spengler 反应得4,5,6,7-四氢噻吩并[3,2-C]吡啶盐酸盐,最后和α-溴代邻氯苯乙酸甲酯反应得外消旋氯吡格雷。
1实验
1.1试剂与仪器
α-溴代邻氯苯乙酸和2-噻吩乙胺为工业品(纯
度>98%),其余试剂均为市售化学纯或分析纯试剂。
高效液相色谱仪(美国Waters 公司),WRS-1A 数字熔点仪(上海物理光学仪器厂),NMR-200型核磁共振波谱仪(美国瓦里安公司),FTIR-8400傅立叶交换红外分光光度计(日本岛津公司)。
1.2实验步骤
1.2.1α-溴代邻氯苯乙酸甲酯的制备
250mL 三口烧瓶中加入α-溴代邻氯苯乙酸24.9g (0.01mol ),100mL 无水甲醇,4g 浓硫酸,搅
拌回流反应5h ,然后蒸馏回收多余的甲醇,冷却后加入50mL 乙醚,20mL 冰水,分层,以5%的碳酸氢钠溶液洗至中性,无水硫酸钠干燥,过滤,有机相减压浓缩,得剩余物约22g (收率83%),可直接用于下一步反应。
1HNMR δ(ppm )(CDCl 3):3.76(3H),5.90(1H),
7.20~7.78(4H)。
1.2.24,5,6,7-四氢噻吩并[3,2-C]吡啶盐酸盐的制备
500mL 三口烧瓶中加入200mL 的乙醇氯化氢
溶液,2-噻吩乙胺20.3g (0.16mol ),5.7g (0.19mol )多聚甲醛,回流反应5h ,浓缩至干,剩余物加无水乙醇重结晶,过滤,烘干得白色固体21.1g (收率
75%)。
1HNMR δ(ppm )(d-DMSO):3.04(2H),3.32(2H),4.22(2H),6.92(1H),7.44(1H),9.81(2H)。
6--
2010年第41卷第5期
《浙江化工》
1.2.3外消旋氯吡格雷硫酸盐的制备
250mL 三口烧瓶中加入甲醇100mL ,4,5,6,7-四氢噻吩并[3,2-C]吡啶盐酸盐8.8g (0.05mol ),碳酸钾13.8g (0.1mol ),搅拌30min ,然后加入α-溴代邻氯苯乙酸甲酯13.2g (0.05mol ),升温至回流,反应6h 。
冷却,过滤,滤液减压浓缩至干,加入50
mL 乙酸乙酯,加热回流,冷却,过滤,滤液滴加5g
浓硫酸,析出固体,过滤,丙酮洗涤,烘干得到白色晶体17.3g (收率82%)。
1HNMR δ(ppm )(D 2O):3.12
(2H),3.62(2H),3.70(3H),4.23(2H),5.72(1H),6.60(1H),7.21(1H),7.40~760(4H)。
2结论
本文合成了外消旋氯吡格雷硫酸盐,总收率达
61.5%(以2-噻吩乙胺计),其IR 谱图和标志样一致,
其产品也可以以盐酸盐、磺酸盐等其他酸的盐存在。
在合成4,5,6,7-四氢噻吩并[3,2-C]吡啶盐酸盐时[9],以盐酸水溶液作溶剂,发现其产物中总有约
30%的2-噻吩乙胺因水解未能反应,导致产物分离
较难;改为本文的方法后产物处理较易。
参考文献:
[1]Mills D C B,Puri R,Hu C J,et a1.Clopidogrel inhibits the binding of ADP analogues to the receptor mediating inhibition of platelet adenylate cyclase [J].Arteriosclerosis and Thrombosis,1992,12:430-436.
[2]Salvi P,Heilmann E,Nurden P,et a1.Clopidogrel:An antithrombotic drug acting on the ADP -dependent activation pathway of human platelets
[J].Clinical and Applied
Thrombosis/Hemostasis,1996,2:35-42.
[3]姜丹.急性缺血性脑血管病早期诊断和对策[J].安徽
医药,2005,9(8):624-626.
[4]Khatri H N,Dehoff B S.Process for preparation of 2-thiophene ethanamine and its conversion to ficlopidine:EP,0439404[P].1991-07-31.
[5]Deseamps M,Radisson J.Preparation of methyl α-[4,5,5,7-tetrahydrothieno [3,2-c]pyrid -5-y1]-2-chlorophenyl acetate:EP,0466569[P].1992-01-15.
[6]Maria A B,Cstarin M,Molnarl M,et a1.New interm ediates for clopidogrel and analogs and process for their preparation:WO,199851681[P].1998-11-19.
[7]Radisson J,Braye E.Preparation of thienylethyland bis (thienylethy1)amines as intermediates for antihacterial agent:EP,0274324[P].1988-07-13.
[8]Boulder E L,Louisville H N K.2-(2-nitrovinyl)thiophene reduction and synthesis of thieno [3,2-C]pyridine derivatives:US,4906756[P].1990-03-06.
[9]Roquettes A B,Toulouse D F.Dextro-rotatory enantiomer of methyl alpha-5-(4,5,6,7-tetrahydro[3,2-c]thienopyridy1)(2-chloropheny1)-acetate and the pharmaceutical compositions containing it:US,4847265[P].1989-07-11.
Synthesis of Racemic Clopidogrel
XU Ke-hui,LIU Pei-pei,LIN Fei-fei,PAN Fan-bing,GUO Hai-chang
(School of Pharmaceutical and Chemical Engineering,Taizhou University,Linhai 317000,China)
Abstract:Racemic clopidogrel was synthesized from 2-(2-thienyl)ethylamine through Pictet Spenglerand reaction to give 4,5,6,7-tetrahydrothieno [3,2-C]pyridine,then reacted with methyl α-bromo -2-chloro -phenylacetate.The overall yield was 61.5%.
Key words:clopidogrel;2-(2-thienyl)ethylamine;methyl α-bromo-2-chloro-phenylacetate;synthesis
7--。