PCR常见问题的精辟总结
- 格式:doc
- 大小:104.50 KB
- 文档页数:16

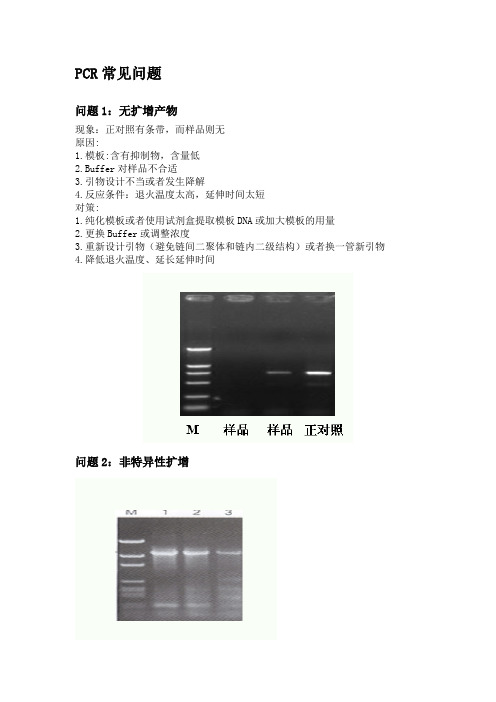
PCR常见问题问题1:无扩增产物现象:正对照有条带,而样品则无原因:1.模板:含有抑制物,含量低2.Buffer对样品不合适3.引物设计不当或者发生降解4.反应条件:退火温度太高,延伸时间太短对策:1.纯化模板或者使用试剂盒提取模板DNA或加大模板的用量2.更换Buffer或调整浓度3.重新设计引物(避免链间二聚体和链内二级结构)或者换一管新引物4.降低退火温度、延长延伸时间问题2:非特异性扩增现象:条带与预计的大小不一致或者非特异性扩增带原因:1.引物特异性差2.模板或引物浓度过高3.酶量过多4.Mg2+浓度偏高5.退火温度偏低6.循环次数过多对策:1.重新设计引物或者使用巢式PCR2.适当降低模板或引物浓度3.适当减少酶量4.降低镁离子浓度5.适当提高退火温度或使用二阶段温度法6.减少循环次数问题3:拖尾现象:产物在凝胶上呈Smear状态。
原因:1.模板不纯2.Buffer不合适3.退火温度偏低4.酶量过多5.dNTP、Mg 2+浓度偏高6.循环次数过多对策:1.纯化模板2.更换Buffer3.适当提高退火温度4.适量用酶5.适当降低dNTP和镁离子的浓度6.减少循环次数问题4:假阳性现象:空白对照出现目的扩增产物原因:靶序列或扩增产物的交*污染对策:1.操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外;2.除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。
所用离心管及加样枪头等均应一次性使用。
3.各种试剂最好先进行分装,然后低温贮存PCR引物设计的黄金法则(转自tiangen)1.引物最好在模板cDNA的保守区内设计。
DNA序列的保守区是通过物种间相似序列的比较确定的。
在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区2.引物长度一般在15~30碱基之间。
引物长度(primer length)常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA 聚合酶进行反应。

PCR反应常见问题汇总(转自tiangen)----PCR常见问题分析及对策1.PCR产物的电泳检测时间一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚致消失。
2.假阴性,不出现扩增条带PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量及,④PCR 循环条件。
寻找原因亦应针对上述环节进行分析研究。
模板:①模板中含有杂蛋白质,②模板中含有Taq酶抑制剂,③模板中蛋白质没有消化除净,特别是染色体中的组蛋白,④在提取制备模板时丢失过多,或吸入酚。
⑤模板核酸变性不彻底。
在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处理,模板核酸提取过程出了毛病,因而要配制有效而稳定的消化处理液,其程序亦应固定不宜随意更改。
酶失活:需更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而导致假阴性。
需注意的是有时忘加Taq酶或溴乙锭。
引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散的常见原因。
有些批号的引物合成质量有问题,两条引物一条浓度高,一条浓度低,造成低效率的不对称扩增,对策为:①选定一个好的引物合成单位。
②引物的浓度不仅要看OD 值,更要注重引物原液做琼脂糖凝胶电泳,一定要有引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无条带,此时做PCR有可能失败,应和引物合成单位协商解决。
如一条引物亮度高,一条亮度低,在稀释引物时要平衡其浓度。
③引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏部分,导致引物变质降解失效。
④引物设计不合理,如引物长度不够,引物之间形成二聚体等。
Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
反应体积的改变:通常进行PCR扩增采用的体积为20ul、30ul、50ul。

PCR实验常见问题、原因分析及其解决方案PCR产物的电泳检测时间,一般为48h以内,有些最好于当日进行检查,大于48h后带型不规则甚至消失。
但有时仍会与到这样那样的问题,影响检测结果的判断,具体归类为以下常见的4点,描述如下:问题一:无扩增产物现象:正对照有条带,而样品则无原因:1、模板:含有抑制物,含量低2、Buffer对样品不合适3、引物设计不当或者发生降解4、反应条件:退火温度太高,延伸时间太短对策:1、纯化模板或者使用试剂盒提取模板DNA或加大模板的用量2、更换Buffer或调整浓度3、重新设计引物(避免链间二聚体和链内二级结构)或者换一管新引物4、降低退火温度、延长延伸时间问题二:非特异性扩增现象:条带与预计的大小不一致或者非特异性扩增带原因:1、引物特异性差2、模板或引物浓度过高3、酶量过多4、Mg2+浓度偏高5、退火温度偏低6、循环次数过多对策:1、重新设计引物或者使用巢式PCR2、适当降低模板或引物浓度3、适当减少酶量4、降低镁离子浓度5、适当提高退火温度或使用二阶段温度法6、减少循环次数问题三:拖尾现象:产物在凝胶上呈Smear状态原因:1、模板不纯2、Buffer不合适3、退火温度偏低4、酶量过多5、dNTP、Mg 2+浓度偏高6、循环次数过多对策:1、纯化模板2、更换Buffer3、适当提高退火温度4、适量用酶5、适当降低dNTP和镁离子的浓度6、减少循环次数问题四:假阳性现象:空白对照出现目的扩增产物原因:靶序列或扩增产物的交叉污染对策:1、操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外;2、除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。
所用离心管及加样枪头等均应一次性使用。
3、各种试剂最好先进行分装,然后低温贮存[ 来源]:实验室之家,以及相关网络知识、转载仅为分享知识,如有侵权请联系删除。
}热文推荐:1、化学分析方法确认和验证指南PDF版全文(新版2018年4月1日实施)2、最全微生物实验室规划设计方案3、谱知识总结篇4、移液器操作六部曲,这些细节很重要化学先生(号码:chemistrysir),化学检测工作者自己的公众号。

普通PCR常见问题及解决对策普通PCR常见问题及解决对策⼀、普通PCR常见问题及解决对策PCR产物的电泳检测时间⼀般为48h以内,有些最好于当⽇电泳检测,⼤于48h 后带型不规则甚致消失。
1. 假阴性,不出现扩增条带PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量及活性④PCR循环条件。
寻找原因亦应针对上述环节进⾏分析研究。
1.1 模板①模板中含有杂蛋⽩质,②模板中含有Taq酶抑制剂,③模板中蛋⽩质没有消化除净,特别是染⾊体中的组蛋⽩,④在提取制备模板时丢失过多,或吸⼊酚。
⑤模板核酸变性不彻底。
在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处理,模板核酸提取过程出了⽑病,因⽽要配制有效⽽稳定的消化处理液,其程序亦应固定不宜随意更改。
1.2 酶失活需更换新酶,或新旧两种酶同时使⽤,以分析是否因酶的活性丧失或不够⽽导致假阴性。
需注意的是有时忘加Taq酶或溴⼄锭。
1.3 引物引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散的常见原因。
有些批号的引物合成质量有问题,两条引物⼀条浓度⾼,⼀条浓度低,造成低效率的不对称扩增,对策为:①选定⼀个好的引物合成单位。
②引物的浓度不仅要看OD值,更要注重引物原液做琼脂糖凝胶电泳,⼀定要有引物条带出现,⽽且两引物带的亮度应⼤体⼀致,如⼀条引物有条带,⼀条引物⽆条带,此时做PCR有可能失败,应和引物合成单位协商解决。
如⼀条引物亮度⾼,⼀条亮度低,在稀释引物时要平衡其浓度。
③引物应⾼浓度⼩量分装保存,防⽌多次冻融或长期放冰箱冷藏部分,导致引物变质降解失效。
④引物设计不合理,如引物长度不够,引物之间形成⼆聚体等。
1.4 Mg2+浓度Mg2+离⼦浓度对PCR扩增效率影响很⼤,浓度过⾼可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚⾄使PCR扩增失败⽽不出扩增条带。
1.5 反应体积的改变通常进⾏PCR扩增采⽤的体积为20ul、30ul、50ul。

PCR技术具有高度的特异性、选择性、灵敏性,而且快速、简便、易于自动化,因此在临床医学工作中受到广泛的重视及合理的应用。
目前在我国许多基层医疗单位均已相继开展P CR研究用应用工作。
PCR技术很简单,原理也很清楚,因此有条件的单位都能较顺利地上马,但毕竟PCR技术是一种新生事物,受到许多条件因素的影响,所以要做得好也不容易。
在PCR技术应用过程中会遇到这样或那样的问题,更甚至会得到与事实完成相反的结果。
为了使我们能够顺利地开展PCR工作,更好地发挥PCR技术的工具作用,我们根据多年来PCR工作总结的经验,对PCR扩增过程中经常出现的一些问题进行简要的总结,提供给广大科研工作者作为参考,抛砖引玉,不足之处欢迎指正。
一、假阳性扩增在实验过程中有时会碰到所检验的样本全部阳性,而且扩增产物电泳条带亮度均一,这是扩增系统受到污染时最典型的一种表现,需仔细检查,排除污染源,一般应从以下步骤着手:⒈试剂污染:厂方提供的试剂或其中的某种组分在出厂或运输、贮存过程中受到污染。
检测方法,可按试剂盒说明书将试剂分装好,直接置于PCR仪中扩增(不加阴性对照,可加适量蒸馏水),便可简单而有效地判断试剂是否已受到污染。
⒉实验室污染:这是最常见的污染源,由于PCR技术可以在几个小时内将模板中某些特异性序列扩增到数百万倍以上,扩弗产物可能在电泳等过程中污染移液器、操作台或随着蒸发所形成气溶胶而污染整个实验室。
扩增产物又是最有效的模板,一旦出现产物污染其污染程度都较严重。
判断方法:可以将实验中最关键步骤如试剂分装、样品处理等转移到一个新的环境中或转移到超净工作台中完成。
当然,所用的移液器、吸咀管(离心管)都应彻底更换。
解决方法:实验室污染是PCR扩增过程中最易出现的现象,而且一旦出现污染,消除污染源又极其困难,往往需对整个实验室及实验器材彻底清洗处理,所以应该以预防为主,实验过程中严格遵守实验基本要求,样品处理与扩增产物及电泳应尽量分开,最好能在两个房间进行。
P C R检测常见问题与解决途径------------------------------------------作者xxxx------------------------------------------日期xxxxPCR检测常见问题与解决途径利用PCR方法检测转基因成分时,经常出现假阳性、假阴性、非特异性扩增和涂抹带。
尤其是假阳性和假阴性可使检测结果得出错误结论,有时可造成严重后果。
为了提高转基因检测的准确性和可靠性,PCR检测应尽量减少假阳性、假阴性、非特异性扩增和涂抹带现象的发生。
一个好的PCR方法不但要求特异性好、灵敏度高、还要求具有高的可重复性、重现性、鲁棒性,尽量减少假阳性、假阴性和非特异性扩增。
本文对PCR检测中出现的假阳性、假阴性、非特异性扩增和涂抹带的原因及其解决方法予以综述。
一、假阳性:(一)假阳性现象假阳性是指检测阴性材料得到阳性结果。
如果一次实验中的几个阴性对照中出现一个或几个阳性结果,提示本次实验中其它标本的检测结果可能有假阳性。
实验中设立的阴性对照可提示有无假阳性结果出现。
(二)造成假阳性的原因1. 样品间交叉污染:样本污染主要有收集样本的容器被污染,或样本放置时,由于密封不严溢于容器外,或容器外粘有样本而造成相互间交叉污染;样本核酸模板在提取过程中,由于吸样枪污染导致标本间污染;2. PCR试剂的污染:主要是由于在PCR试剂配制过程中,由于加样枪、容器、双蒸水及其它溶液被PCR核酸模板污染。
3. PCR扩增产物污染:这是PCR反应中最主要最常见的污染问题。
因为PCR产物拷贝量大(一般为1013拷贝/ml),远远高于PCR检测数个拷贝的极限,所以极微量的PCR产物污染,就可形成假阳性。
4. 气溶胶污染:在空气与液体面摩擦时就可形成气溶胶,在操作时比较剧烈地摇动反应管,开盖时、吸样时及污染进样枪的反复吸样都可形成气溶胶而污染。
据计算一个气溶胶颗粒可含48000拷贝,因而由其造成的污染是一个值得特别重视的问题。
PCR常见问题总汇PCR产物的电泳检测时间一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚致消失。
假阴性,不出现扩增条带PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量及,④PCR循环条件。
寻找原因亦应针对上述环节进行分析研究。
模板:①模板中含有杂蛋白质,②模板中含有Taq酶抑制剂,③模板中蛋白质没有消化除净,特别是染色体中的组蛋白,④在提取制备模板时丢失过多,或吸入酚。
⑤模板核酸变性不彻底。
在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处理,模板核酸提取过程出了毛病,因而要配制有效而稳定的消化处理液,其程序亦应固定不宜随意更改。
酶失活:需更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而导致假阴性。
需注意的是有时忘加Taq酶或溴乙锭。
引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散的常见原因。
有些批号的引物合成质量有问题,两条引物一条浓度高,一条浓度低,造成低效率的不对称扩增,对策为:①选定一个好的引物合成单位。
②引物的浓度不仅要看OD值,更要注重引物原液做琼脂糖凝胶电泳,一定要有引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无条带,此时做PCR有可能失败,应和引物合成单位协商解决。
如一条引物亮度高,一条亮度低,在稀释引物时要平衡其浓度。
③引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏部分,导致引物变质降解失效。
④引物设计不合理,如引物长度不够,引物之间形成二聚体等。
Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
反应体积的改变:通常进行PCR扩增采用的体积为20ul、30ul、50ul。
或100ul,应用多大体积进行PCR扩增,是根据科研和临床检测不同目的而设定,在做小体积如20ul 后,再做大体积时,一定要模索条件,否则容易失败。
PCR常见问题的精辟总结--耶鲁大学作者:佚名来源:耶鲁大学时间:2007-9-5 Troubleshooting for PCR and multiplex PCR Troubleshooting discussion is based on the PCR protocol as described in the table below. All reactions are run for 30 cycles. Component volume final concentration1.autoclaved ultra-filtered water (pH 7.0)20.7µL-2.10x PCR Buffer*2.5µL1x3.dNTPs mix (25 mM each nucleotide)0.2µL200 µM (each nucleotide)4.primer mix (25 pmoles/µL each primer)0.4µL0.4 µM (each primer)5.Taq DNA polymerase (native enzyme)0.2µL1 Unit/25 µL6.genomic DNA template (100 ng/µL)1.0µL100 ng/25 µL* The 10x PCR buffer contains: 500 mM KCl; 100 mM Tris-HCl (pH 8.3); 15 mM MgCl2(the final concentrations of these ingredients in the PCR mix are: 50 mM KCl; 10mM Tris-HCl; 1.5 mM MgCl2).问题解决:1. I get (many) longer unspecific products. What can I do?Decrease annealing timeIncrease annealing temperatureDecrease extension timeDecrease extension temperature to 62-68º CIncrease KCl (buffer) concentration to 1.2x-2x, but keep MgCl2concentration at 1.5-2mM.Increase MgCl2 concentration up to 3-4.5 mM but keep dNTP concentrationconstant.Take less primerTake less DNA templateTake less Taq polymeraseIf none of the above works: check the primer for repetitive sequences(BLAST align the sequence with the databases) and change the primer(s)Combine some/all of the above2. I get (many) shorter unspecific products. What can Ido?Increase annealing temperatureIncrease annealing timeIncrease extension timeIncrease extension temperature to 74-78º CDecrease KCl (buffer) concentration to 0.7-0.8x, but keep MgCl2concentration at 1.5-2mMIncrease MgCl2 concentration up to 3-4.5 mM but keep dNTP concentrationconstantTake less primerTake less DNA templateTake less Taq polymeraseIf none of the above works: check the primer for repetitive sequences(BLAST align the sequence with the databases) and change the primer(s)Combine some/all of the above3. Reaction was working before, but now I can't get any product.Make sure all PCR ingredients are taken in the reaction (buffer, template, Taq, etc)Change the dNTP solution (very sensitive to cycles of thawing and freezing, especially in multiplex PCR)If you just bought new primers, check for their reliability (bad primer synthesis ?)Increase primer amountIncrease template amountDecrease annealing temperature by 6-10º C and check if you get any product. If you don't, check all your PCR ingredients. If you do get products (including unspecific ones) reaction conditions as described above. Combine some/all of the above4. My PCR product is weak. Is there a way to increase the yield?Gradually decrease the annealing temperature to the lowest possible.Increase the amount of PCR primerIncrease the amount of DNA templateIncrease the amount of Taq polymeraseChange buffer (KCl) concentration (higher if product islower than 1000bp or lower if product is higher than 1000bp) Add adjuvants. Best, use BSA (0.1 to 0.8 µg/µL final concentration). You can also try 5% (v/v, final concentration) DMSO or glycerol.Check primer sequences for mismatches and/or increase the primer length by5 nucleotidesCombine some/all of the above5. My two primers have very different melting temperatures (Tm) but I cannot change their locus. What can I do to improve PCR amplification?An easy solution is to increase the length of the primer with low Tm. If you need to keep the size of the product constant, add a few bases at the 3' end. If size is not a concern, add a few bases at either the 3' or the 5' end of that primer.6. I have a number of primer pairs I would like to use together. Can I run a multiplex PCR with them?. How?Very likely, yes.Try amplify all loci seaprately using the same PCR program. If one of the primer pairs yields unspecific products,keep the cycling conditions constant and change other parameters as mentioned above (#1 and #2). Mix equimolar amounts of primers and run the multiplex reaction either in the same cycling conditions or by decreasing only the annealing temperature by 4º C.If some of the loci are weak or not amplified, read below !!7. How many loci can I amplify in multiplex PCR at the same time?Difficult to say. The author has routinely amplified from 2 to 14 loci.Literature describes up to 25 loci or so.8. One or a few loci in my multiplex reaction are very weak or invisible.How can amplify them?The first choice should be increasing the amount of primer for the "weak" loci at the same time with decreasing the amount of primer for all loci that can be amplified. The balance between these amounts is more important than the absolute values used !!.Check primer sequences for primer-primer interactions9. Short PCR products in my multiplex reaction are weak. How can I improve their yield?Increase KCl (buffer) concentration to 1.2x-2x, but keep MgCl2concentration at 1.5-2mMDecrease denaturing timeDecrease annealing time and temperatureDecrease extension time and temperatureIncrease amount of primers for the "weak" loci while decreasing the amountfor the "strong" loci.Add adjuvants. Best, use BSA (0.1 to 0.8 µg/µL final concentration). Youcan also try 5% (v/v, final concentration) DMSO or glycerolCombine some/all of the above10. Longer PCR products in my multiplex reaction are weak. How can I improve their yield?Decrease KCl (buffer) concentration to 0.7-0.8x, butkeep MgCl2 concentration at 1.5-2mM Increase MgCl2 concentration up to 3-4.5 mM but keep dNTP concentration constant.Increase denaturing timeIncrease annealing timeDecrease annealing temperatureIncrease extension time and temperatureIncrease amount of primers for the "weak" loci while decreasing the amount for the "strong" loci Add adjuvants. Best, use BSA (0.1 to 0.8 µg/µL final concentration). You can also try 5% (v/v, final concentration) DMSO or glycerol Combine some/all of the above11. All products in my multiplex reaction are weak. How can I improve the yield?Decrease annealing time in small steps (2º C)Decrease extension temperature to 62-68º CIncrease extension timeIncrease template concentrationIncrease overall primer concentrationAdjust Taq polymerase concentrationChange KCl (buffer) concentration, but keep MgCl2concentration at 1.5-2mMIncrease MgCl2 concentration up to 3-4.5 mM but keep dNTP concentration constant. Add adjuvants. Best, use BSA (0.1 to 0.8 µg/µL final concentration). You can also try 5% (v/v, final concentration) DMSO or glycerol Combine some/all of the above12. Unspecific products appear in my multiplex reaction. Can I get rid of them somehow?If long: increase buffer concentration to 1.2-2x, but keep MgCl2 concentration at 1.5-2mMIf short: decrease buffer concentration to 0.7-0.9x, but keep MgCl2concentration at 1.5-2mMGradually increase the annealing temperatureDecrease amount of templateDecrease amount of primerDecrease amount of enzymeIncrease MgCl2 concentration up to 3-4.5 mM but keep dNTP concentration constant。