超高能软强子作用的ChouCYang模型
- 格式:pdf
- 大小:145.77 KB
- 文档页数:6

物理标准模型物理标准模型是物理学领域中的一个重要理论框架,它对自然界的基本粒子和相互作用做出了最基础的描述。
这个模型由几个部分组成,包括粒子物理、核与强相互作用、弱相互作用、电磁相互作用以及相对论与引力。
1.粒子物理粒子物理是研究基本粒子的学科,这些粒子包括电子、质子、中子、光子、夸克、轻子等。
粒子物理的标准模型将这些粒子分类为不同的族,每一族都有相似的属性和行为。
这个模型预测了各种粒子的存在和性质,以及对它们之间相互作用的规律。
2.核与强相互作用核与强相互作用是描述夸克和胶子等粒子在原子核中如何相互作用的理论。
夸克是构成质子和中子等强子的基本单元,而胶子则是传递强相互作用的光子。
核与强相互作用的标准模型预测了各种强子的性质和相互作用方式,以及它们在原子核中的行为。
3.弱相互作用弱相互作用是描述弱玻色子和弱矢量玻色子如何相互作用的。
弱相互作用涉及的粒子有中微子、电子和夸克等。
弱相互作用的标准模型预测了这些粒子的性质和相互作用方式,以及它们如何通过弱相互作用进行转化。
4.电磁相互作用电磁相互作用是描述光子如何传递电磁力的过程。
电磁相互作用涉及的粒子有电子、质子、光子等。
电磁相互作用的标准模型预测了这些粒子的性质和相互作用方式,以及光子的行为和性质。
5.相对论与引力相对论与引力是描述时空结构、质能关系以及引力如何产生和传递的理论。
相对论由爱因斯坦提出,它预测了物体在高速运动状态下会发生时空弯曲,引力则是由于物体之间的质量引起的时空弯曲现象。
相对论与引力的标准模型预测了引力的作用方式和性质,以及如何与其他基本相互作用进行耦合。
物理标准模型是一个非常成功的理论框架,它解释了大量实验现象并预测了新的物理效应。
然而,这个模型仍然存在一些未解决的问题和局限性,例如暗物质、暗能量以及量子引力等问题,这些问题的解决需要更深入的研究和探索。

波尔模型高三知识点波尔模型是物理学中描述原子结构的理论模型,由丹麦物理学家尼尔斯·波尔于1913年提出。
该模型基于经典力学,有效地描述了原子中电子的能级和能量转换。
一、波尔模型的基本假设波尔模型的基本假设是:1. 电子围绕原子核旋转,并只能在特定轨道上运动;2. 电子在特定轨道上运动时,不会辐射能量,也不会损失能量;3. 电子能量只能取离散的特定值,称为能级。
二、原子结构的主要组成部分根据波尔模型,原子结构主要由以下几个组成部分构成:1. 原子核:位于原子的中心,带正电荷,质量较大;2. 电子壳层:围绕原子核旋转的电子路径,根据能级不同可分为K层、L层、M层等;3. 电子能级:描述电子在轨道上的能量状态,能级越高,电子对原子核的束缚越弱;4. 能级跃迁:电子从一个能级跃迁到另一个能级时,会吸收或释放特定频率的光子能量。
三、波尔模型的应用和局限性波尔模型为我们理解和解释原子结构、光谱现象等提供了重要的理论基础。
然而,该模型并不能完全解释一些实验现象,例如复杂原子的谱线结构和电子自旋等。
在实际应用中,我们通常使用量子力学的理论来更加准确地描述和计算原子结构和性质。
四、波尔模型的实验验证与发展波尔模型提出后,经过一系列实验验证,其基本思想得到了支持,但也遇到了一些困难。
随着科学技术的进步,量子力学的发展逐渐取代了波尔模型,为我们提供了更为精确的原子结构描述。
五、拓展知识:量子力学的影响与应用相较于波尔模型,量子力学能够更精确地描述原子的行为和相互作用,对于高精度计时、量子计算、量子通信等领域有着重要的应用价值。
通过量子理论的研究,科学家们揭示了微观世界的奇妙规律,为我们对于宇宙的认知带来了新的突破。
六、总结波尔模型作为早期原子结构研究的里程碑,为我们打开了研究原子世界的大门。
虽然波尔模型在某些方面有其局限性,但为后续科学研究和发展奠定了基础,让我们更加深入地认识了原子的奥秘。
随着科学不断进步,我们对于原子结构的理解也会不断深化,为人类社会的发展做出更大的贡献。
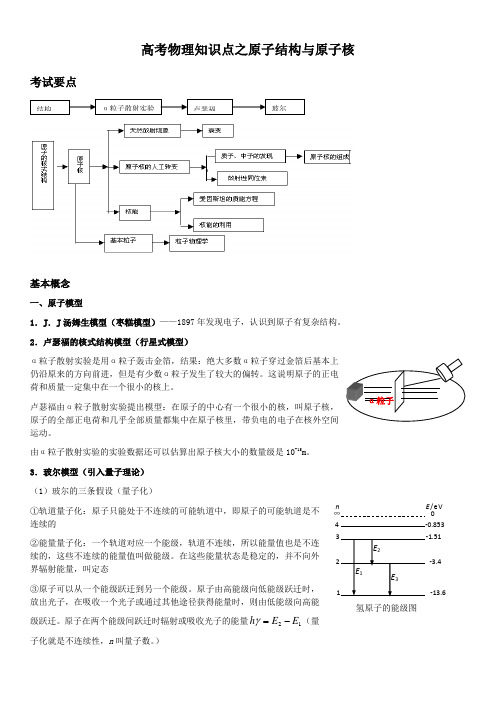
高考物理知识点之原子结构与原子核考试要点基本概念一、原子模型1.J .J 汤姆生模型(枣糕模型)——1897年发现电子,认识到原子有复杂结构。
2.卢瑟福的核式结构模型(行星式模型)α粒子散射实验是用α粒子轰击金箔,结果:绝大多数α粒子穿过金箔后基本上仍沿原来的方向前进,但是有少数α粒子发生了较大的偏转。
这说明原子的正电荷和质量一定集中在一个很小的核上。
卢瑟福由α粒子散射实验提出模型:在原子的中心有一个很小的核,叫原子核,原子的全部正电荷和几乎全部质量都集中在原子核里,带负电的电子在核外空间运动。
由α粒子散射实验的实验数据还可以估算出原子核大小的数量级是10-15m 。
3.玻尔模型(引入量子理论) (1)玻尔的三条假设(量子化)①轨道量子化:原子只能处于不连续的可能轨道中,即原子的可能轨道是不连续的②能量量子化:一个轨道对应一个能级,轨道不连续,所以能量值也是不连续的,这些不连续的能量值叫做能级。
在这些能量状态是稳定的,并不向外界辐射能量,叫定态③原子可以从一个能级跃迁到另一个能级。
原子由高能级向低能级跃迁时,放出光子,在吸收一个光子或通过其他途径获得能量时,则由低能级向高能级跃迁。
原子在两个能级间跃迁时辐射或吸收光子的能量12E E h -=γ(量子化就是不连续性,n 叫量子数。
)α粒子散射实验卢瑟福玻尔结构α粒子氢原子的能级图n E /eV∞ 0 1 -13.62 -3.43 4 -0.853 E 1E 2E 3(2)从高能级向低能级跃迁时放出光子;从低能级向高能级跃迁时可能是吸收光子,也可能是由于碰撞(用加热的方法,使分子热运动加剧,分子间的相互碰撞可以传递能量)。
原子从低能级向高能级跃迁时只能吸收一定频率的光子;而从某一能级到被电离可以吸收能量大于或等于电离能的任何频率的光子。
(如在基态,可以吸收E ≥13.6eV的任何光子,所吸收的能量除用于电离外,都转化为电离出去的电子的动能)。
(3)玻尔理论的局限性。

波尔原子模型波尔原子模型是关于原子结构的一个重要理论模型,是由丹麦物理学家尼尔斯·波尔于1913年提出的。
该模型成功地解释了许多原子的现象和性质,为后续的原子理论研究奠定了基础。
本文将介绍波尔原子模型的基本原理、发展和应用。
波尔原子模型的基本原理是:原子由中央带电核和围绕核运动的电子组成。
核质量集中在原子核中,电子质量相对较小,运动在核外的轨道上。
根据量子力学的理论,电子只能存在于一定能量的轨道上,并且在跃迁时会发射或吸收特定能量的光子。
波尔通过对氢原子光谱进行研究,发现了许多规律。
他提出了以下几条假设:电子在不同的轨道上运动时,具有不同的能量;电子在轨道上保持稳定的运动,不会辐射能量;电子在不同轨道间跃迁时,会吸收或发射光子,并且吸收或发射的光子能量与电子跃迁的能级差相关。
根据这些假设,波尔建立了波尔原子模型。
他认为,电子在距离原子核较远的轨道上运动时,电子的能量较高;而在距离原子核较近的轨道上运动时,电子的能量较低。
当电子从一个低能级的轨道跃迁到一个高能级的轨道时,会吸收能量;当电子从一个高能级的轨道跃迁到一个低能级的轨道时,会发射能量。
波尔原子模型的提出对解释氢原子的光谱非常有效。
根据波尔的理论,氢原子的光谱可以通过电子的跃迁来解释。
当电子处于基态(最低能级)时,不吸收外部能量,不发射光线,处于稳定状态。
当电子从基态跃迁到激发态时,吸收了特定能量的光子。
而当电子从激发态跃迁回基态时,会发射特定能量的光子。
根据这些跃迁能级和光子能量的关系,可以准确地预测氢原子光谱线的位置和强度。
波尔原子模型的发展并不止于氢原子。
其后续的研究证明了波尔原子模型对其他元素的适用性,特别是单电子离子。
对于多电子原子,波尔原子模型的简化假设无法解释其复杂的光谱现象,因此后来的研究发展出了更加复杂的模型,如量子力学的多电子原子理论。
然而,尽管波尔原子模型存在一些局限性,它仍然为我们理解原子结构和性质提供了一个重要的框架。


Gaussian软件应用——高精度能量模型第七章高精度能量模型前两章中,我们讨论了不同理论方法和基组的计算精度,也讨论了各自的优缺点,本章讨论得到非常精确结果的方法.高精度模型的建立,能够是关于能量的计算精度达到2kcal/mol的差距.一般的, 达到这样的精度需要一个庞大的QCISD(T)计算,甚至对于小分子的处理,其运算量也是惊人的.G2,CBS-4,CBS-Q方法是包括了一系列采用特别方法处理的计算的组合,可以提供更为精确的结果.7.1 预测热化学我们主要讨论的是原子化能,电子亲和势,离子化能和质子亲和能.原子化能原子化能是分子与组成分子的原子的能量差,如对于PH2,其原子化能为E(P)+2(EH())-R(PH2)例7.1 文件e7_01 PH2的原子化能采用B3LYP/6-31G(d)优化几何构型,计算零点能(矫正因子0.9804),用B3LYP/6-31+G(d,p)计算能量.得到的原子化能为148,3kcal/mol,实验值为144,7,误差3.6kcal/mol电子亲和势电子亲和势电子亲和势指体系增加一个电子后能量的变化,计算方法为中性分子和其阴离子的能量差.同上例中计算方法得到的PH2电子亲和势为1.24eV,实验值1.26eV, 误差0.02eV,大约0.5kcal/mol离子化能离子化能指体系减少一个电子的能量的变化,计算方法为中性分子和其阳离子的能量差距.同上两例计算方法得到的离子化能为9.95eV,实验值9,82eV,误差-0.13eV 约-2.9kcal/mol.质子亲和能质子亲和能为体系增加一个质子后的能量变化,计算方法为分子与在其基础上增加一个质子的体系的能量差距.同上例计算方法得到的质子亲和能为185.9kcal/mol,实验值为187.1kcal/mol,差距1.2kcal/mol.7.2 理论模型的评价理论模型一般采用上面的热力学数据来评价7.3 G2分子基(Molecule Set)以及缺陷及对缺陷的解释G2分子基是在55个原子化能,38个离子化能,25个电子亲和势和7和质子亲和能的基础上发展的.这个分子基有很多优点,使得其能够得到精确的热力学结果* 热力学数据一般是很难模拟的,误差产生于模型假设中的缺陷.* 实验值也是有误差的* 该分子基包含了大量的原子* 该分子基包含了大量的特殊体系,如离子,开壳层体系等其缺点是,* 其所处理的分子体系小,推广到大的体系是必须要小心* 不是所有的键型都支持的,比如不包括环状分子,没有C-F键* 只能研究前两周期原子,推广到其他原子,如过渡金属可能会有问题* 由于其产生于非常精确的热力学数据,其本身是武断的,甚至对于一些一二周期原子的双原子分子不能全部得到精确结果这一点本身很重要,因为从一小部分分子的某个热力学数据得到的理论模型在应用上必须小心.7.4 理论模型的相对精确性通过对半经验(AM1),HF方法,MP(MP2),DFT(B3LYP, SVWN)等理论方法的比较,统计,有如下结论* 最精确的方法是B3LYP/6-311+G(3df,2df,2p)//B3LYP/6-31G(d),注意其表示用后一种方法优化结构,用前一种方法计算能量及性质.这不是最昂贵的计算方法.* 一般的,由中等级理论进行优化,再进行高等级计算的方法比完全采用高等级方法的结果要好.采用高等级的计算,不能够为几何优化带来更为精确的结果.* 基组大小的增加对于几何优化是不必须的,只是对能量的精确描述上有必要* 半经验方法与Hartree-Fock方法比较,其绝对平均误差要小,但最大误差要大, 说明其经验值中包含了一些电子相关,但对于一些体系的处理明显不好,比如离子化能和质子化能的计算* 在B3LYP水平上进行的计算结果,在几何优化上有明显的优势.这些结论显示,* 如果可能,使用B3LYP/6-31G(d)进行几何构型和零点能计算,使用B3LYP的最大基组进行能量计算* 一些研究者推荐使用HF/6-31G(d)零点能和热力学矫正,对于一些大的体系,进行,HF优化和频率分析,然后进行B3LYP/6-31G(d)能量计算比使用B3LYP/6-31G(d)要有效率* 当B3LYP/6-31G(d)太昂贵而无法进行优化和频率分析时,可以使用HF/3-21G进行优化和单点能及矫正 * 使用AM1进行优化的体系,进行B3LYP的单点能计算也能明显提高最终结果的精度.7.5 组合方法一些组合方法用于得到更为精确的结果.这里讨论Gaussian-n方法和完全基组方法(CBS)Gaussian-1 和Gaussian-2理论Gaussian-1和Gaussian-2方法是在优化好的结构上对能量进行修正.下面是Gaussian-1 (G1)方法的处理步骤第一步: 采用HF/6-31G(d)产生初始的几何构型和频率分析得到零点能ZPE, 矫正因子0.8929第二步: 从上一步的优化结果开始,采用MP2(Full)/6-31G(d)进行几何优化.所得几何构型用于后面的计算第三步: 计算基态能量Ebase,在上一步得到的几何结构上采用MP4/6-311G(d,p) 计算.得到的数值在后面进一步矫正第四步: 增加弥散函数,采用MP4/6-311G+(d,p)计算基态能量,与上一个数值比较得到dE+第五步: 增加高级极化函数,采用MP4/6-311G(2df,p)计算基态能量,与第三步的数值比较得到dE2df.如果该数值为正,则设该项为零第六步: 采用QCISD(T)/6-311G(d,p)计算基态能量,差值为dEQCI第七步: 矫正第六步的结果,dEHLC=-0.00019na + -0.00595nb,其中na,nb是处于alpha和beta自旋状态的电子的数目,这样,就得到的G1能量EG1 = Ebase + dE+ + dE2df + dEQCI + dEHLC + ZPE这样得到的EG1和QCISD(T)/6-311+G(2df,p)得到的结果近似,但速度要快的多.G2方法在G1的基础上,增加处理步骤第八步: 运行MP2/6-311+G(3df,2p)能量计算,dEG2 = dE+2df - dE+ - dE2df + dE3d2p将G1方法中的2df项进行修正,由于所需要的MP2计算可以在前面找到,最终的dEG2的计算可以表示为 dEG2 = E(8) - E(5,MP2) - E(4,MP2) + E(3,MP2)其中数字代表进行的步骤,后面的方法为该步骤中该理论的能量值.第九步:将G1中的dEHLC修正,增加0.00114nb,记为dHLCG2能量为EG2 = EG1 + dEG2 + dHLC例7.5 文件e7_05 PH3质子化能(PA)的G2计算结果如下方法G1 G2 G2(MP2) 实验PA 186.10 186.14 186.80 187.1CPU 682.4 829.1 607.5其中G2(MP2)方法是在G2基础上的更为廉价的方法.差距均在2kcal/mol以下.下面是对三种方法的统计结果方法平均绝对误差最大误差G1 1.53 7.4G2 1.21 4.4G2(MP2) 1.58 6.3G2方法是最精确的,也是最昂贵的方法,G2(MP2)在三种方法中是较为经济而且结果也较好的.注意随体系的增加,G2方法的特点就更为明显完全基组方法(Complete Basis Set Motheds, CBS)这个名字本身代表了对从热力学头算方法的最大误差来源-对基组的切断的修正.和G2理论一样,该方法的能量也是有一系列的修正得到的.计算方法基于如下的原理* 对总能量的连续的贡献随着微扰的等级升高而降低,比如对于氧分子体系解离能的计算,精确到0.001Hartree,用SCF方法需要6个描述,而MP2方法需要3个,更高等级的微扰只需要2个.CBS方法基于此而随着计算理论等级的增加采用较小的基组.* CBS方法采用成对中性轨道扩张的渐进收敛,从有限元基组外推建立完全基组 CBS方法一般包括大基组的HF计算,中等基组的MP2计算,以及一个中等略低等级基组的高精度计算,见下表CBS-4 CBS-Q几何优化HF/3-21G(d) MP2/6-31G(d)ZPE(校正因子) HF/3-21G(d) (0.91671) HF/6-31G (0.91844)SCF能量HF/6-311+G(3d2f,2df,p) HF/6-311+G(3d2f,2df,2p)二级修正MP2/6-31+G MP2/6-311+G(3d2f,2df,2p)CBS外推>=5个构造>=10高等级校正MP4(SDQ)/6-31G MP4(SDQ)/6-31+G(d(f),d,f)QCISD(T)/6-31+G经验校正单或双电子高等级校正双电子高阶校正自旋校正自旋校正,对钠的核校正CBS-4比其他两个方法要便宜,另外的CBS方法是CBS-APNO,更加精确也更加昂贵例7.6 文件e7_06 PH3质子化能的CBS计算结果如下方法CBS-4 CBS-Q 实验PA 189.25 186.24 187.1CPU 256.7 708.7两个方法都得到很好的结果.当得到同样精度结果时,当然便宜的方法是好的.下面是CBS和G2方法的统计结果方法绝对平均误差最大误差相对CPU时间PH3 F2CO SiF4CBS-4 1.98 7.0 1.0 1.0 1.0G2(MP2) 1.58 6.3 2.4 10.3 11.5CBS-Q 1.01 3.8 2.8 8.4 12.7G2 1.21 4.4 3.2 25.9 59.1能够达到误差小于2kcal/mol的精确标准,CBS-4是最便宜的.CBS-Q有比G2好的结果,同时也便宜很多练习练习7.1 文件7_01a~d CBS-4的热力学数据计算水的四个热力学数据,在原子化能和电子亲和势方面有很精确的结果,其他两项也符合很好练习7.2 文件7_02a~c 臭氧的氯化解离练习6.9中讨论过该反应,当时没有得到好的结果,下面是高精度计算的结果dH CPUG2 -33.1 6172.3CBS-4 -41.4 1109.4CBS-Q -38.4 3384.4实验-39.1很显然CBS-Q方法得到了很好的结果。
强子物理的标准模型强子物理是研究物质内部组成及其相互作用的学科,而强子物理的标准模型(SM)是描述了强子物理的基本理论框架。
它涵盖了我们对基本粒子、相互作用力以及它们之间关系的理解。
本文将介绍强子物理的标准模型的主要组成部分和基本原理。
一、标准模型的基本粒子标准模型将所有基本粒子分为两类:费米子和规范玻色子。
费米子是具有半整数自旋的粒子,包括了夸克和轻子。
夸克是构成强子的基本组成元素,而轻子则包括了电子、中微子和它们的反粒子。
规范玻色子则是具有整数自旋的粒子,用于描述粒子之间的相互作用力。
标准模型中的规范玻色子包括了光子、带电弱玻色子(W和Z玻色子)以及八种胶子。
二、强子的组成强子由夸克组成,而夸克有六种不同的"味道":上夸克、下夸克、粲夸克、奇夸克、顶夸克和底夸克。
不同的夸克组合形成了不同类型的强子,例如质子和中子都是由上夸克和下夸克组成的核子。
夸克之间的相互作用是通过胶子的交换来实现的,这就是强相互作用。
三、背景20世纪60年代至70年代初,物理学家们提出了强相互作用的理论和基础,但是直到20世纪70年代晚期,标准模型的框架才被正式提出。
标准模型成功地解释了在强相互作用下的粒子行为,并预测了一些粒子的存在,后来这些粒子通过实验证实。
标准模型得到了广泛认同,并被视为粒子物理学的基石之一。
四、标准模型的量子色动力学标准模型的量子色动力学(QCD)是描述夸克之间相互作用的理论。
QCD认为夸克具有色荷,它们通过交换胶子进行相互作用。
QCD预测了在高能量尺度下,夸克和胶子之间的相互作用非常强大,这就解释了为什么夸克在自由态很少被观测到,而总是以强子的形式存在。
五、标准模型的对称性与破缺标准模型具有一些重要的对称性。
其中包括规范对称性、手性对称性和味对称性。
规范对称性保证了标准模型的规范玻色子(如光子和胶子)的无质量性质。
手性对称性描述了夸克和轻子的左手和右手部分之间的相互转换。
味对称性指的是夸克之间的"味道"变换。
蘑菇模型物理知识点总结蘑菇模型的基本结构包括一个巨大的质子核和一圈环绕着核的轻子。
核内部的质子和中子被认为是由夸克组成的,它们之间通过强力相互作用相互结合,形成核子。
轻子则是通过电磁相互作用和弱力作用相互结合,被束缚在核周围。
蘑菇模型的物理知识点主要涉及到核子内部的夸克结构、强相互作用、量子色动力学、核子结构等方面。
下面将对这些知识点逐一加以说明。
1. 核子内部的夸克结构蘑菇模型假设质子和中子都是由夸克组成的。
夸克是一种基本粒子,它具有一定的电荷(+2/3或-1/3)、色荷(红、绿、蓝)和自旋。
根据夸克结构,质子由两个上夸克和一个下夸克组成,而中子由两个下夸克和一个上夸克组成。
这一假设得到了大量实验数据的支持,如电子散射实验、高能质子-核对撞实验等都证实了核子内部存在夸克。
2. 强相互作用蘑菇模型是建立在强相互作用的基础上的。
强相互作用是一种非常强大的相互作用力,它主要负责核子内部夸克之间的相互作用。
强相互作用的载体是一种称为胶子的粒子,它通过色荷相互作用将夸克结合在一起。
利用量子色动力学理论可以对强相互作用进行定量的描述,并且能够解释许多核子物理现象,如强子共振、夸克喷注等。
3. 量子色动力学量子色动力学是研究强相互作用的理论,它将夸克的颜色概念引入到了物理学中。
根据量子色动力学理论,强相互作用的载体是一种称为胶子的粒子,它具有三种不同的色荷。
夸克和胶子之间的相互作用通过交换胶子实现,这种相互作用是非常强大的,因此导致了核子内部的相对稳定。
4. 核子结构蘑菇模型通过描述质子和中子的结构,从而揭示了核子的内部结构。
在蘑菇模型中,核子被认为是由夸克组成的,夸克之间通过强相互作用相互结合在一起。
而轻子则是通过电磁相互作用和弱力相互结合在一起,形成了核子的外部结构。
这种模型不仅能够解释核子的稳定性,还能够预言核子在高能物理实验中的行为。
总结来说,蘑菇模型是一种用来描述质子和中子内部结构的物理模型。
它包括了核子内部的夸克结构、强相互作用、量子色动力学等物理知识点。
原子模型及特点及应用实例目前公认的原子模型主要有经典原子模型、量子力学原子模型和分子轨道理论等。
下面分别介绍这几种原子模型的特点及应用实例。
一、经典原子模型(也称“平面模型”):经典原子模型是由英国科学家托姆逊于1898年提出的,认为原子是由带正电的球体(核)和带负电的电子云组成。
这个模型的特点包括:1. 原子由带正电的球体(核)和带负电的电子云组成;2. 电子云呈球形分布;3. 原子是不可分割的基本粒子。
经典原子模型虽然存在一定的局限性,但仍有一些应用,例如:1. 解释了原子中电子和质子的存在;2. 为后续的量子力学原子模型的发展奠定了基础。
二、量子力学原子模型:量子力学原子模型是由德国科学家薛定谔于1926年提出的,它建立在量子力学理论的基础上,描述了原子的结构和性质。
这个模型的特点包括:1. 具有能级概念:原子内的电子围绕原子核以不同的能级存在,能级越高离核越远;2. 具有波粒二象性:电子在原子中既具有粒子性又具有波动性;3. 具有不确定性原理:不能同时准确确定电子的位置和动量。
量子力学原子模型具有广泛的应用,例如:1. 解释了能级跃迁和光谱现象的规律,为光谱分析提供了理论基础;2. 解释了原子中电子的排布规律,为元素周期表的建立提供了依据;3. 描述了化学键的形成和性质,为化学反应理论提供了基础;4. 通过计算机模拟,可以预测和设计新材料的性质。
三、分子轨道理论:分子轨道理论是对分子中电子状态的描述,是量子力学原子模型的拓展。
这个理论的特点包括:1. 将分子中的电子视为在整个分子空间中运动的波函数,而不是局限于原子核附近;2. 通过波函数叠加来得到分子轨道,分为成键轨道和反键轨道;3. 分子轨道能级和电子分布直接影响分子的性质和反应。
分子轨道理论在化学研究和工业生产中有广泛的应用,例如:1. 解释了分子的结构、性质和反应规律,为有机合成、配位化学等领域提供了理论指导;2. 预测了分子的光谱性质,为红外光谱、紫外光谱等的解释和应用提供了理论支持;3. 研究了分子间的相互作用,为化学反应动力学、反应速率等问题的研究提供了理论依据。
第 55 卷第 1 期2024 年 1 月中南大学学报(自然科学版)Journal of Central South University (Science and Technology)V ol.55 No.1Jan. 2024基于团簇微观结构分析的离子电活性聚合物驱动特性王红,杨亮,杨延宁(延安大学 物理与电子信息学院,陕西 延安,716000)摘要:首先,对离子交换膜吸附水分子微观过程进行分析,并结合团簇结构揭示离子电活性聚合物的传质动力学特性;其次,基于溶胀理论研究团簇受力情况,依据几何变形特点和变形传质机理建立物理模型;最后,对得到的驱动模型进行验证和分析讨论。
研究结果表明:本文模型所得结果和实验结果较吻合。
含水量对离子交换膜团簇通道的形成有重要影响,阳离子的迁移以及水分子运动是离子交换膜驱动的主控因素。
随着阳离子浓度和水分子浓度增加,静水压力、渗透压力以及静电压力均逐渐增大,渗透压力对离子电活性聚合物弯曲变形起主导作用。
关键词:离子聚合物金属复合材料;团簇微观结构;软体机器人;驱动特性中图分类号:TB381 文献标志码:A 文章编号:1672-7207(2024)01-0106-10Actuation characteristics of ionic electroactive polymers based oncluster microstructure analysisWANG Hong, YANG Liang, YANG Yanning(School of Physics and Electronic Information, Yan'an University, Yan'an 716000, China)Abstract: Firstly, analysis of the microscopic process of water molecule adsorption by ion-exchange membranes was carried out and the mass transfer kinetics of ion-electrically active polymers in the context of cluster structure was revealed. Secondly, physical model based on the geometrical deformation characteristics and deformation mass transfer mechanism was established, and the force on the clusters was studied based on the dissolution theory. Finally, the obtained model was experimentally validated and analytically discussed. The results show that the proposed actuation model result matches well with the experimental result. The water content has an important influence on the formation of cluster channels in ion exchange membranes, and the migration of cations and the收稿日期: 2023 −06 −18; 修回日期: 2023 −10 −08基金项目(Foundation item):国家自然科学基金资助项目(52365069);陕西省教育厅科学研究计划项目(23JK0730);延安大学博士科研启动基金资助项目(YDBK2021-09,YDBK2023-09) (Project(52365069) supported by the National Natural Science Foundation of China; Project(23JK0730) supported by Scientific Research Program of Department of Education of Shaanxi Province; Projects(YDBK2021-09, YDBK2023-09) supported by the PhD Research Startup Foundation of Yan'an University)通信作者:杨亮,博士,副教授,从事微纳机械系统、柔性智能材料、仿生人工肌肉以及机器人技术研究;E-mail :*************************.DOI: 10.11817/j.issn.1672-7207.2024.01.009引用格式: 王红, 杨亮, 杨延宁. 基于团簇微观结构分析的离子电活性聚合物驱动特性[J]. 中南大学学报(自然科学版), 2024, 55(1): 106−115.Citation: WANG Hong, YANG Liang, YANG Yanning. Actuation characteristics of ionic electroactive polymers based on cluster microstructure analysis[J]. Journal of Central South University(Science and Technology), 2024, 55(1): 106−115.第 1 期王红,等:基于团簇微观结构分析的离子电活性聚合物驱动特性movement of water molecules are the main controlling factors leading to the actuation of ion exchange membranes. The hydrostatic, osmotic and electrostatic pressures increase with the increase of the cation and water molecule concentrations, and the osmotic pressure plays dominant role in the bending and deformation of ion-electrically active polymers.Key words: ionic polymer-metal composites; cluster microstructures; soft robotics; actuation characteristics大力发展机器人产业对于促进国民经济具有重要意义。