朗格汉斯细胞组织细胞增生症诊疗指南【2019版】
- 格式:docx
- 大小:25.72 KB
- 文档页数:6
![朗格汉斯细胞组织细胞增生症[指南]](https://uimg.taocdn.com/c4e21558ff4733687e21af45b307e87100f6f844.webp)
朗格汉斯细胞组织细胞增生症郎格罕细胞组织细胞增生症(Langenhans cell histiocytosis,LCH),原称组织细胞增生症,是一组原因未明的组织细胞增殖性疾患。
传统分为三种临床类型,即莱特勒西韦综合征(Litterer-Siwe病,简称L-S病),汉-薛-柯综合征(Hand-Schuller-Christian病,简称H-S-C病)及骨嗜酸肉芽肿(eosinphilic granuloma of bone ,EGB)。
病因未明,近年来研究发现多与体内免疫调节紊乱有关。
本病发病率估计1/20万-1/200万.主要发生在婴儿和儿童,也见于成人甚至老人.不少报告提到男性患者居多.嗜酸性肉芽肿是一种孤立性的组织细胞的非肿瘤性质的异常分化。
嗜酸性肉芽肿是郎罕氏细胞增多症的一种表现,以前称为组织细胞增多症X。
嗜酸性肉芽肿多发生于5-10岁的儿童,侵犯部位为骨骼和肺。
这占郎罕氏细胞增多症病例的60-80%。
黑人本症极少见。
嗜酸性肉芽肿可见于颅骨,下颔骨、脊柱和长管骨。
男女发生率比2:1。
嗜酸性肉芽肿的临床表现为:局部疼痛、肿胀,血沉升高。
嗜酸性肉芽肿病灶的放射线表现无特殊性,不同的部位,表现不同。
颅骨的病损表现为内外颅骨板层不规则的锋利的破坏,形成“斜边缘”,(Beveled edge),骨盆的缺损边界多模糊。
脊椎缺损多发生于椎体。
发生在长骨,则多位于股干,干骺端的髓腔中份。
病损可以造成骨内、骨膜的反应。
CT扫描及MRI 对于明确病变髓腔内的范围及对皮质骨的破坏程度有价值。
影像学诊断需与尤文氏肉瘤、骨肉瘤、转移瘤和化脓性骨炎鉴别。
大体组织检查、嗜酸性肉芽肿是软的、肉芽状、胶质状的组织。
颜色灰红、褐色或者褚黄色。
显微镜下观,嗜酸性肉芽肿内大量郎罕氏细胞。
这些郎罕氏细胞来源于单核细胞和髓腔内的树突状细胞。
电镜下,这些细胞与郎罕氏细胞一样都含有浆内的颗粒状小体,Birbeck’s颗粒。
镜下尚可见数量不等的淋巴细胞,多核砼性细胞、嗜酸性细胞和巨细胞。


朗格汉斯细胞组织细胞增生症及其治疗朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,简称LCH)是一组少见的瘤样病变,其年发病率约为4/1000 000。
过去因其病因不明而称为组织细胞增生症X,近年通过电镜及免疫组化研究认为,肿瘤组织细胞中可发现朗格汉斯细胞的表面标记和典型的Birbeck颗粒,其组织细胞实际上是变异的郎格汉斯细胞(LC),故现称为郎格汉斯细胞组织细胞增生症,是发生在骨组织的反应性、非肿瘤性、增生性疾病。
多见于20岁以下青少年,5~10岁之间为发病高峰,男性略多于女性。
LCH以朗格汉斯细胞增生引起的骨病变为主,按照发病年龄和临床特点,一般分为3型,即:①勒-雪病(Abt-Letterer-Siwe disease,LS),②韩-薛-柯病(Hand-Schüller-Christian disease,HSC),③骨嗜酸性粒细胞肉芽肿(Eosinophilic granuloma of bone,EGB)或称单发性嗜酸性粒细胞肉芽肿。
韩-薛-柯病多见于2~4岁儿童,又称为多发性嗜酸性粒细胞肉芽肿。
勒-雪病以婴儿多发,青少年发病较少,可很快致死。
该2型的临床特点、治疗和预后详见儿童口腔颌面部肿瘤章节。
本节重点阐述颌骨嗜酸性粒细胞肉芽肿。
骨嗜酸性粒细胞肉芽肿由Finzi于1929年首次报告,后由Jaffe(1940)命名为骨嗜酸性粒细胞肉芽肿。
本病临床少见,在国内5所口腔医学院校病理科统计的20122例颌面部肿瘤中仅61例,约占各类肿瘤的3%。
1.病因和发病机制2.临床表现EBG是LCH中最常见的一型,主要表现为慢性炎症,进展较慢,病程长。
多见于儿童或青少年,偶见于老年人,有报道4岁以下的儿童占34%。
男女之比为2∶1。
全身骨骼均可发病,主要侵犯长骨、颅骨(额骨>顶骨>颞骨>枕骨)、肋骨、盆骨等。
可单发,也可多发。
发生于颌骨的嗜酸性粒细胞肉芽肿以单发者常见,多发者约占25%。


老年朗格汉斯细胞组织细胞增生症3例诊治分析张旭;赵翌【摘要】目的探讨老年朗格汉斯细胞组织细胞增生症患者的临床特征及治疗方法.方法收集3例经病理学证实的老年朗格汉斯细胞组织细胞增生症患者的临床资料.本文3例老年患者均以淋巴结及内脏器官侵犯为主,无皮疹,1例低热.1例行化疗,1例行放化疗,1例行放疗;观察他们的临床特征,并根据RECIST标准评价治疗疗效、CTCAE v3.0进行安全性评估、观察总体生存期.结果行放/化疗后患者的肿块缩小,生存期延长,最长26个月.主要副作用为放化疗后骨髓抑制.结论老年朗格汉斯组织细胞增生症的患者,以淋巴结及内脏器官侵犯为主,放/化疗是一种较好的治疗选择.%Objective To investigate the clinical features and treatment of the elderly Langerhans cell histiocytosis patients. Methods We collected 3 elderly clinical data, which confirmed by pathology of Langerhans cell histiocytosis. The lesions mainly affected the lymph nodes and internal organs, no rash, and one case of low fever. One patient received chemotherapy, one patient received radiotherapy and chemotherapy, one patient radiotherapy. We observed their clinical features. We evaluated treatment efficacy according to the RECIST criteria, safety assessment by CTCAE v3.0,and observed the patients's overall survival. Results The mass reduced after radiotherapy /chemotherapy, overall survival longer, up to26 months; the main side effect was bone marrow suppression after radiotherapy and chemotherapy. Conclusion The lesions mainly affectedthe lymph nodes and internal organs in the elderly Langerhans cellhistiocytosis patients, radiotherapy/ chemotherapy maybe is a better treatment option to them.【期刊名称】《大连医科大学学报》【年(卷),期】2013(035)002【总页数】4页(P163-166)【关键词】朗格汉斯细胞组织细胞增生症;临床特征;治疗【作者】张旭;赵翌【作者单位】大连医科大学附属第一医院肿瘤科,辽宁大连116011【正文语种】中文【中图分类】R733.1朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis, LCH)是一种倾向于肿瘤性病变的疾病,容易累及皮肤、内脏、骨等组织导致死亡。


朗格汉斯细胞组织细胞增生症(Langerhanscellhistiocytosis,LCH)【概述】朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)是一组原因未明的以属于单核巨噬细胞系统的特定树突细胞和网状细胞增生为共同特点的疾病,以往曾称为组织细胞增生症X(histiocytosis X)。
这类疾病较少见,但在儿童组织细胞增生症中是最常见的一种。
根据累及部位的不同,将其分为3种类型:①孤立性嗜酸性肉芽肿;② Hand-Schuller-Christian病;③ Letterer-Siwe病。
正常情况下人体内有一定数量的但分布局限的朗格汉斯细胞,其主要出现在表皮和黏膜的复层鳞状上皮基底层内,也可见于胃肠黏膜上皮、真皮、淋巴结、脾脏等。
但当朗格汉斯细胞出现克隆性增生时,就表现出本病。
【流行病学】本病发病率估计为(0.5--5)/100万,儿童发病率每年约为1/250万。
LCH可发生于任何年龄,但大多发生在婴儿和儿童,诊断的高峰年龄在1--3岁,也可见于成人,甚至老人。
以男性多见,男女比例为3.7:1。
孤立性嗜酸性肉芽肿多见于大龄儿童和青年(常近30岁),发病高峰出现在5--10岁;Hand-Schuller-Christian 病多见于2--6岁的儿童;Letterer-Siwe病一般见于3个月--3岁的男童。
【病因】病因不明。
有人认为与接触石棉、儿童期未接种牛痘有关;偶见婴儿出生时已有本病,又很早死亡,故有人认为本病可能与宫内感染有关;也有人认为发病可能与感染有关,其理由是抗生素治疗有时能使病情好转或延长缓解时间,甚至“治愈”。
但在病变组织中一般均未能证实有细菌、真菌或病毒存在。
成人肺LCH几乎均与吸烟有关,因为部分患者戒烟后症状即可缓解。
关于LCH属于良性增生性疾病还是恶性肿瘤,鉴于多数病例病程急剧,常致死亡,大量增生的组织细胞形态又像是恶性细胞,少数病例最后可演变成单核细胞白血病,因此世界卫生组织(WHO)造血和淋巴组织肿瘤(2001)及骨和软组织分类(2002)把LCH明确定位为肿瘤性疾病。

成人朗格汉斯细胞组织细胞增生症的治疗进展成人朗格汉斯细胞组织细胞增生症的治疗进展概述成人朗格汉斯细胞组织细胞增生症(Adult Langerhans cell histiocytosis,ALCH)是一种罕见的多系统疾病,主要由成人骨髓内发育异常的朗格汉斯细胞引发。
该疾病临床上以骨骼损害、肺部病变和皮肤损害为主要特征。
本文将重点介绍成人朗格汉斯细胞组织细胞增生症的治疗进展。
药物治疗1. 糖皮质激素糖皮质激素是主要的治疗药物之一。
以泼尼松为代表的糖皮质激素疗法可以抑制朗格汉斯细胞的增殖和活性,并减轻症状。
然而,长期使用可能引发一系列副作用,如骨质疏松、高血压和免疫抑制等。
2. 化疗药物阿霉素是一种化疗药物,可用于成人朗格汉斯细胞组织细胞增生症的治疗。
它通过抑制朗格汉斯细胞的增殖和活性,从而减轻症状和改善疾病进程。
然而,阿霉素的使用会引发严重的副作用,如肝脏损伤、骨髓抑制和感染等。
3. 靶向治疗药物最近的研究表明,靶向治疗药物在处理成人朗格汉斯细胞组织细胞增生症中起到了积极的作用。
噻吗班尼是一种针对朗格汉斯细胞增生的BRAF V600E突变的靶向治疗药物。
该药物与RAS/RAF/MEK/ERK信号通路有关,通过抑制该信号通路的活性来达到抑制朗格汉斯细胞增生的效果。
在临床试验中,噻吗班尼显示出了显著的疗效,改善了患者的症状和生存率。
其他治疗方法1. 造血干细胞移植对于患有反复复发或晚期疾病的患者,造血干细胞移植可以考虑作为治疗方法。
该方法通过将患者的造血干细胞替换为健康的供者干细胞,达到修复异常造血系统的作用,提高患者的生存率。
2. 局部治疗对于局限于骨骼或皮肤等特定部位的病变,局部治疗是常用的方法。
常见的局部治疗方法包括手术切除、放射治疗和局部激素注射等。
这些方法可以减轻局部病变的症状,但对于全身性疾病效果有限。
结语成人朗格汉斯细胞组织细胞增生症是一种罕见而复杂的疾病,目前尚无完全治愈的方法。
药物治疗是常用的治疗手段,但药物的长期使用和副作用限制了其应用。

朗格汉斯细胞组织细胞增生症分类朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)是一种罕见的多系统疾病,主要由朗格汉斯细胞(Langerhans cells)异常增生引起。
朗格汉斯细胞是一种专门从骨髓祖细胞发展而来的树突状细胞,正常情况下主要分布在淋巴结、皮肤、肺部和骨骼等组织中,起到调节免疫和炎症反应的作用。
然而,在LCH患者中,朗格汉斯细胞异常增生,并在体内形成肿块,进而导致不同器官的功能损害。
根据病变的部位和表现方式,LCH通常被分为三个主要的临床亚型:单系统疾病、多系统疾病和散发性疾病。
1.单系统疾病:单系统疾病是指LCH仅影响一个器官或系统,约占所有LCH患者的50-60%。
常见的单系统病变包括骨骼(骨骺、骨质破坏)、皮肤(单个或多个皮肤病变)、淋巴结(淋巴结肿大)和中枢神经系统(颅骨骨髓窦、脑膜血管炎等)。
单系统疾病通常具有较好的预后,大多数患者经过治疗后疾病得到控制。
2.多系统疾病:多系统疾病是指LCH累及两个或两个以上的非相邻器官或系统。
约有30-40%的LCH患者呈现多系统疾病表现。
多系统疾病通常涉及骨骼、皮肤、淋巴结、脾脏、肝脏等多个器官,病情较为严重。
疾病的严重程度和进展速度因患者而异,治疗方案也较为复杂。
多系统疾病可能会导致器官功能损害和全身性症状,如贫血、发热、体重下降等。
3.散发性疾病:散发性疾病是指LCH广泛累及全身多个器官或系统,包括骨骼、皮肤、淋巴结、脾脏、肝脏、中枢神经系统等。
这是LCH最严重和最少见的一种表现方式,约占所有LCH患者的10%以下。
散发性疾病在诊断和治疗上常常具有挑战性,需要综合多个专科的团队进行协作管理。
此外,根据LCH的病程和预后划分,还可以将LCH分为急性、亚急性和慢性三种类型。
急性型病程进展迅速,累及多个器官,预后较差。
亚急性型病程进展相对缓慢,患者多为儿童和青少年,预后一般较好。
慢性型进展缓慢,预后相对良好。
儿童肺朗格汉斯细胞组织细胞增生症的诊断及治疗完整版儿童肺朗格汉斯细胞组织细胞增生症(pPLCH)是一种罕见的弥漫性囊性肺疾病。
与成人PLCH通常为肺单器官受累不同,pPLCH通常为多系统LCH的一个组成部分。
虽然肺部受累相比其他器官的受累的预后较好,但是受到多系统受累的影响,气胸、呼吸衰竭等并发症也可危机生命。
朗格汉斯细胞组织细胞增生症(LCH)是一种罕见的组织细胞疾病,其特点为各种组织和器官中朗格汉斯细胞的异常增生和浸润。
目前认为该病是一种炎性髓系瘤。
LCH可分为单系统型及多系统型,单系统性LCH较为多见,骨是单系统型LCH的最常见受累部位,而多系统型LCH可包括皮肤、骨骼、淋巴结、胸腺、肺、肝、脾、骨髓、中枢神经系统;其中包括肝、脾或骨髓受累的“高危组”患者的死亡风险较高,过去肺部受累时也被分入高危组,但近期认为肺部受累比其他器官受累的患者的预后较好,但是受到多系统受累的影响,气胸、呼吸衰竭等并发症也可危机生命。
肺朗格汉斯细胞组织细胞增生症(PLCH)通常发生于成人,并且通常为单系统受累,主要与吸烟有关,与之不同,儿童PLCH(pPLCH)比较少见,且通常不会单独受累,为多系统LCH的一个组成部分。
本文则主要对pPLCH进行介绍。
表1 儿童与成人PLCH的异同pPLCH通常表现为干咳、呼吸急促,还可能出现胸痛、乏力、发热、体重减轻等。
出现肺大疱并引起气胸,疼痛、呼吸困难可能是首发症状,气胸在儿童时较少发生。
由于许多肺部受累的患者仅有轻微或无症状,而肺外体征和症状较为明显,因此,无论既往病史或检查结果如何,在诊断有LCH后,需要对患者进行胸片检查,以评估是否累及肺部。
若以肺部症状为首发症状的儿童患者,也需要评估其他部位是否受累。
检查是否出现皮疹、疼痛、肿胀、发热、耳鸣、多尿及神经症状等。
诊断PLCH是一种罕见疾病,除了临床症状和影像学检查外,还需要组织病理学诊断及免疫表型检查。
影像学儿童PLCH与成人在影像学上表现相似,胸片最常见双侧间质浸润,呈网状结节状,伴支气管增厚,偶尔也可见囊性改变和气胸。
可治性罕见病—朗格汉组织细胞增生症一、疾病概述朗格汉组织细胞增生症( Langerhans cell histiocytosis.LCH).原称组织细胞增多症X,是一组由未成熟树突细胞活化增殖并且异常聚集为主要特征的罕见疾病。
可发生于任何年龄段,以婴幼儿和儿童常见,儿童发病率约2~10/100万,男性多见,白种人发病率较其他人种高;儿童LCH患者的总体病死率约为15%,永久性后遗症发生率为30%~40%[l]。
该病的发病机制尚不明确,该病为免疫系统失调导致的反应性增生还是肿瘤性一直存在争议。
二、临床特征LCH临床表现差异较大,可长期仅表现为轻微皮疹,单一部位溶骨性损害,无明显临床进展。
也可表现为多个重要脏器受累,病情进展迅速,甚至导致死亡。
Haupt等[1]于2013年报道,该病最常累及脏器为骨骼(80%),皮肤(33%)和垂体(25%),其他累及器官主要有肝、脾、造血系统、肺部(15%)、淋巴结(5%~10%)以及中枢神经系统。
骨骼是最常见的受累器官。
在大约80%的LCH患者中都存在骨损害,最常见的受累部位为颅骨(27%),其次为大腿骨(13%).下颌骨(11%),骨盆(10%),颅骨损害多可累及软脑膜,但严重的颅内受累非常罕见。
常见的颅骨损害以眶周骨,颞骨(特别是乳突)为主;眶周骨病变可导致眼球凸出。
乳突病变可同时出现中耳炎。
皮疹常为就诊的首发症状。
皮肤的病变可表现为脂溢性、湿疹样、脓疱性和结节性皮炎等,典型皮疹为散在分布的红色丘疹伴有中央溃疡区形成,溃疡结痂后,触诊存在棘手感。
单纯的皮肤受累有60%的机会可自行缓解。
但需要严密的病情监测,剩下的40%可出现复发,并进展成为多系统LCH。
对于存在眶周骨、筛骨、颧骨、颞骨受累的患者,发生中枢神经系统受累的可能性(25%)要高于无受累患者。
尿崩症是中枢神经系统受累的LCH患者的初期表现;在儿童中,单独出现的尿崩症和垂体增强MRI上增粗的垂体柄,最常见的诊断为LCH、生殖细胞瘤、淋巴瘤。
朗格汉斯组织细胞增生症的诊治进展作者:张鼎赵斌来源:《医学信息》2019年第19期摘要:朗格汉斯组织细胞增生症(LCH)是一种少见的、病因尚不明确的单核-巨噬细胞异常增生性疾病,其临床表现具有多样性,累及多个系统,且发病机制复杂,目前主要认为是炎性髓样肿瘤,治疗上仍以联合化疗为主,随着靶向药物及骨髓移植应用逐渐增多,其并发症及不良反应需进一步解决。
对于单系统受累LCH 患者预后较佳,临床报道治愈病例较多。
而多系统受累LCH患者预后不佳,病死率高。
本文就近年来有关LCH的发病机制、临床表现、诊断与治疗及预后进展进行综述。
关键词:朗格汉斯组织细胞增生症;单核-巨噬细胞异常增生;炎性髓样肿瘤中图分类号:R55 ; ; ; ; ; ; ; ; ; ; ; ; ; ; ; ; ; ;文献标识码:A ; ; ; ; ; ; ; ; ; ; ; ; ; ; ; ; ;DOI:10.3969/j.issn.1006-1959.2019.19.012文章编号:1006-1959(2019)19-0035-04Progress in the Diagnosis and Treatment of Langerhans Cell HistiocytosisZHANG Ding1,ZHAO Bin2(1.Fujian University of Traditional Chinese Medicine,Fuzhou 350108,Fujian,China;2.Xiamen Hospital of Traditional Chinese Medicine,Xiamen361009,Fujian,China)Abstract:Langerhans cell histiocytosis (LCH) is a rare mononuclear-macrophage dysplasia with unclear etiology. Its clinical manifestations are diverse, involving multiple systems, and the pathogenesis is complex. At present, it is mainly considered to be an inflammatory myeloid tumor. The treatment is still based on combined chemotherapy. With the increasing application of targeted drugs and bone marrow transplantation, its complications and adverse reactions need to be further resolved. The prognosis of LCH patients with single system involvement is better, and more cases are reported in clinical reports. The multi-system involvement of LCH patients has a poor prognosis and a high mortality rate. This article reviews recent advances in the pathogenesis, clinical manifestations, diagnosis and treatment, and prognosis of LCH.Key words:Langerhans cell histiocytosis;Mononuclear-macrophage dysplasia;Inflammatory myeloid tumor朗格汉斯细胞是(langerhans cell)起源于骨髓前体细胞的巨噬细胞(组织细胞),是单细胞系列的一部分。
先天自愈性朗格汉斯组织细胞增生症怎样
*导读:本文向您详细介绍先天自愈性朗格汉斯组织细胞增生症的治疗方法,治疗先天自愈性朗格汉斯组织细胞增生症常用的西医疗法和中医疗法。
先天自愈性朗格汉斯组织细胞增生症应该吃什么药。
*先天自愈性朗格汉斯组织细胞增生症怎么治疗?
*一、西医
*1、治疗
可自愈,不必特殊治疗,但应密切观察。
*2、预后
皮损可迅速自行消退,身体及智力发育正常。
可自愈。
*温馨提示:上面就是对于先天自愈性朗格汉斯组织细胞增生症怎么治疗,先天自愈性朗格汉斯组织细胞增生症中西医治疗方法的相关内容介绍,更多更详尽的有关先天自愈性朗格汉斯组织细胞增生症方面的知识,请关注疾病库,也可以在站内搜索“先天自愈性朗格汉斯组织细胞增生症”找到更多扩展资料,希望以上内容对大家有帮助!。
60.朗格汉斯细胞组织细胞增生症
概述
朗格汉斯细胞组织细胞生多症(Langerhans cell histiocytosis,LCH)是一种组织细胞疾病,旧称“组织细胞增生症X”。
2017 年版WHO 组织细胞疾病和巨噬-树突细胞系肿瘤分类标准中将其与Erdheim-Chester 病(ECD)共同分为L 组。
目前认为是LCH 是一种炎性髓系肿瘤。
病因和流行病学
目前发现约50%LCH 患者的病变组织存在着BRAF V600E 突变,在BRAF 野生型患者中,33%~50%可以发现MAP2K1(编码MEK1 的基因)突变或丝裂原活化蛋白激酶(MAPK)信号通路中其他基因突变(如ARAF 和ERBB3 等)。
BRAF V600E 突变可发生在造血细胞的不同发育阶段,这也会影响LCH 的临床表现和分型。
例如,如果突变发生于骨髓干祖细胞阶段,临床多表现为多系统高危型,而仅发生于朗格汉斯细胞阶段时,则多表现为单系统低危型。
因此,目前认为LCH 是一种以MAPK 信号通路激活为主要特征的克隆性血液系统肿瘤,属于炎性髓系肿瘤。
LCH 的年发病率估计为0.5/100 000~5.4/100 000,男性稍多,本病常见于儿童,成人LCH 发病率低。
临床表现
LCH 的临床表现多种多样。
病情从轻至重差异很大,因此容易被误诊和漏诊。
LCH 的临床表现主要包括:
1.发热热型不规则,可呈周期性或者持续性高热。
下丘脑受累患者可以出现中枢性体温调节异常。
2.皮疹主要分布于躯干、头皮和发际。
初起为淡红色丘疹,然后可以为出血性或湿疹样皮脂溢出样皮疹,继而结痂,结痂脱落后留有白斑。
3.口腔及眼耳鼻喉病变牙龈肿胀、牙齿松动,突眼,顽固性中耳炎伴外耳道皮疹为典型表现。
4.中枢性尿崩症垂体、下丘脑甚至其他部位中枢神经系统病变,少数患者出现神经精神症状。
5.呼吸道症状咳嗽,重者憋喘、发绀,甚至反复气胸发作。
肺部体征通常不明显。
6.肝脾可以肿大,可以出现黄疸、肝功能异常、肝功能衰竭和门脉高压,最终患者可死于肝硬化和肝功能衰竭。
7.消化道症状消化道累及较少见,可以表现为息肉或腺瘤样改变,通常不出现临床症状,病变弥漫时可能出现腹痛、腹泻和低蛋白血症等。
8.造血系统淋巴结可见肿大,骨髓受累时可以出现贫血、白细胞和血小板计数异常。
9.骨质损害颅骨、四肢骨、脊柱、骨盆等可有疼痛及肿块,可以发生病理性骨折。
眼眶骨质病变所致突眼为儿童患者典型表现之一。
辅助检查
1.血常规无特异性改变。
以不同程度贫血多见,多为正细胞正色素性贫血。
重症患者可见血小板降低。
白细胞分类中,仅1/6 的儿童患者嗜酸粒细胞>4%,因此难以肯定血中嗜酸粒细胞与嗜酸细胞肉芽肿的关系。
2.血液生化肝酶和胆管酶增高可能代表肝脏受累,严重时可以出现类似肝硬化的异常表现。
尿崩患者可有血钠升高,尿液渗透压低于血浆渗透压。
3.炎症指标可以出现血沉增快和 C 反应蛋白(CRP)升高,这些指标可能反映出疾病的活动性。
4.内分泌指标生长激素缺乏(GHD)、性激素缺乏、促肾上腺皮质激素(ACTH)缺乏和促甲状腺激素(TSH)缺乏等都是常见异常。
5.骨质评价X 线和C T(如全身低剂量C T)均可作为首选方法。
全身骨质均可以发生破坏,病变特征为溶骨性骨质破坏。
扁骨病灶为虫蚀样至巨大缺损,颅骨巨大缺损可呈地图样。
脊柱多为椎体破坏,呈扁平椎。
但椎间隙不变窄。
长骨多为囊状缺损,无死骨形成。
MRI 和P ET-CT 也是可以选择的检查方法,其优势在于可以同时发现骨质外病变,并且对治疗效果进行评价。
由于存在假阳性可能,不宜将同位素骨显像作为单一的评价手段。
6.肺部检查高分辨CT 可见肺部弥漫性网状或点网状阴影,也可见局限或颗粒状阴影,类似于粟粒性肺结核。
严重病例可见肺气肿或蜂窝状肺囊肿、纵隔气肿、气胸或皮下气肿。
也可做肺功能以及支气管肺泡灌洗液检查,支气管肺泡灌洗液中CD1a 阳性细胞>5%支持诊断。
胸腺增大在儿童患者中也可见到,尸检证实肿大的胸腺是组织细胞浸润的结果。
7.其他影像学检查可以采用B 超检查颈部甲状腺肿物和腹腔脏器,颅内病变、尤其是垂体情况适合采用MRI 进行检查,典型表现可见垂体后亮点缺失、垂体柄增粗。
8.P ET-CT 有助于评价疾病全身受累范围,受累脏器会有不同程度放射性浓聚表现。
诊断方法
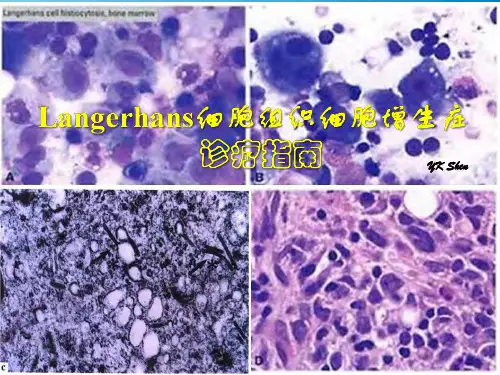
病理诊断是LCH 诊断的金标准。
LCH 的典型病理表现:光镜下可见分化较好的组织细胞增生。
此外,可见泡沫样细胞、嗜酸粒细胞、淋巴细胞、浆细胞和多核巨细胞。
慢性病变中可见大量含有多脂质性的组织细胞和嗜酸粒细胞,形成嗜酸细胞肉芽肿,增生中心可有出血和坏死。
除了上述光镜下特点外,确诊还需要免疫组化检查,巨细胞的CD68、CD1a、S100 及langerin(CD207)均为阳性。
电镜检查可见朗格汉斯巨细胞,这种细胞是一种体积较大的单个核细胞,直径可达13μm,胞体不规则。
胞浆中可见被称为朗格汉斯颗粒或者Birbeck 颗粒的分散的细胞器,颗粒长190~
360nm,宽33nm,末端可呈泡沫样扩张,形态如网球拍。
细胞核不规则,常呈扭曲状,核仁明显,多为1~3 个。
约50%的LCH 患者存在BRAF V600E 基因突变。
鉴别诊断
主要需要与其他组织细胞疾病相鉴别,如ECD 和未定类树突细胞肿瘤。
1.E CD 是一种罕见的非朗格汉斯细胞组织细胞增生症。
在2017 年版WHO 组织细胞疾病和巨噬-树突细胞系肿瘤分类标准中,将其与朗格汉斯细胞组织细胞增生症(LCH)共同分为L 组。
病理是鉴别诊断的金标准。
ECD 病变组织中CD1a、CD207 均为阴性,同时电镜下无LCH 特征性的Birbeck 颗粒。
但由于LCH。