美国环保局 EPA 试验 方法美国环保局 EPA 试验 方法 9045dSoil and Waste pH
- 格式:pdf
- 大小:29.78 KB
- 文档页数:5


银离子含量检测标准
银离子含量的检测标准通常由各个国家或地区的标准化组织制定。
以下是一些常见的银离子含量检测标准:
1. 国际标准化组织(ISO):ISO 11581-1:2009金属化学分析方法-银、铱和铜含量检测-电感耦合等离子体质谱法(ICP-MS)分析
2. 美国环保局(EPA):EPA方法200.7/200.8/6020A-金属分析方法-银含量检测-电感耦合等离子体质谱法(ICP-MS)分析
3. 欧洲标准化组织(EN):EN 13805-食品中常见元素含量的测定-ICP-MS法-银含量检测
4. 中华人民共和国国家标准:GB/T 5009.121-食品安全国家标准-饮用水中微量元素的测定-电感耦合等离子体质谱法(ICP-MS)分析-银含量检测
需要注意的是,不同国家或地区的标准可能存在一定的差异,具体的检测方法和限量标准应根据当地的法律法规或相关行业标准确定。

Internet9071B - 1Revision 2April 1998METHOD 9071Bn-HEXANE EXTRACTABLE MATERIAL (HEM) FOR SLUDGE, SEDIMENT, AND SOLID SAMPLES 1.0SCOPE AND APPLICATION1.1Method 9071 may be used to quantify low concentrations of oil and grease in soil,sediments, sludges, and other solid materials amenable to chemical drying and solvent extraction with n-hexane. “Oil and grease” is a conventional pollutant under 40 CFR 401.16 and generally refers to substances, including biological lipids and mineral hydrocarbons, that have similar physical characteristics and common solubility in an organic extracting solvent. As such, oil and grease is an operationally defined parameter, and the results will depend entirely on the extracting solvent and method of extraction. Method 9071 employs n-hexane as the extraction solvent with Soxhlet extraction and the results of this method are appropriately termed “n-hexane extractable material (HEM).” Section 1.2 lists the type of materials that may be extracted by this method. In the context of this method, “HEM” is used throughout this method and for operational purposes, may be considered synonymous with “oil and grease” within the limitations discussed below.1.2Specifically, Method 9071 is suitable for extracting relatively non-volatile hydrocarbons,vegetable oils, animal fats, waxes, soaps, greases, biological lipids, and related materials. 1.3Method 9071 is not recommended for measuring materials that volatilize at temperatures below 85E C. Petroleum fuels from gasoline through #2 fuel oil may be partially lost during the solvent removal process.1.4 Some crude oils and heavy fuel oils may contain materials that are not soluble in n-hexane, and recovery of these materials may be low.2.0SUMMARY OF METHOD2.1 A representative portion of wet (as received) waste is acidified with concentrated HCl and chemically dried with magnesium sulfate or sodium sulfate. Magnesium sulfate monohydrate is used to dry acidified sludges as it will combine with 75% of its own weight in water in forming MgSO C 7H O. Anhydrous sodium sulfate is used to dry soil and sediment samples. 4 2 2.2After drying, the HEM is extracted with n-hexane using a Soxhlet apparatus. The n-hexane extract is then distilled from the extract and the HEM is desiccated and weighed. 2.3 When necessary, a separate sample portion is evaluated for percent solids, and the dry weight fraction may be used to calculate the dry-weight HEM concentration of the soil, sediment,or waste.3.0DEFINITIONS3.1n-Hexane extractable material (HEM, oil and grease): Material that is extracted from a sample using n-hexane and determined by this method. This material includes relatively non-volatile hydrocarbons, vegetable oils, animal fats, waxes, soaps, greases, and related matter.3.2Refer to Chapter One for additional definitions.4.0INTERFERENCES4.1 This method is entirely empirical, and duplicate results having a high degree of precision can be obtained only by strict adherence to all details. The rate of cycling and time of extraction in the Soxhlet apparatus must be consistent and length of time required for drying and cooling extracted materials must be the same in order to generate consistent results. It is important that the procedures be performed as directed due to the varying solubilities of the different greases and heavy mineral oils.4.2Solvents, reagents, glassware, and other sample-processing hardware may yield artifacts that could affect the results. All solvents and reagents used in the analysis should be demonstrated to be free from interferences by processing a method blank with each analytical batch. Specific selection of reagents, solvent washes, or purification of solvents may be required. Use of plastic measuring devices, and/or plastic tubing attachments must be avoided.4.3Glassware should be cleaned by washing with hot tap water with detergent, rinsing with tap water and reagent water, and rinsing with solvent. Glassware may also be baked at 200-250E C for 1 hour. Boiling flasks that are used to contain the extracted residues may be dried in an oven at 105-115E C and stored in a desiccator until used. Depending on the project DQOs, strict adherence to the washing and handling procedures cited above may not be necessary as long as the laboratory can demonstrate that alternative cleaning procedures yield acceptable method performance and meet method blank acceptance criteria.4.4 A gradual increase in weight may result due to the absorption of oxygen; a gradual loss of weight may result due to volatilization. Extracted residues should be maintained in a desiccator during cooling and prior to weighing. Extracted residues should be weighed as soon as possible after cooling.4.5The presence of non-oily extractable substance such as sulfur compounds, organic dyes, and chlorophyll, may result in a positive bias. For the purpose of this method, all materials extracted and retained during this procedure are defined as HEM.5.0SAFETY5.1The toxicity or carcinogenicity of each reagent used in this method has not been precisely determined; however, each chemical should be treated as a potential health hazard. Exposure to these chemicals should be reduced to the lowest possible level. It is suggested that the laboratory perform personal hygiene monitoring of each analyst that uses this method. This monitoring should be performed using Occupational Safety and Health Administration (OSHA) or National Institute of Occupational Safety and Health (NIOSH) approved personal hygiene monitoring methods. Results of this monitoring should be made available to the analyst.5.2n-Hexane has been shown to have increased neurotoxic effects over other hexanes and some other solvents. OSHA has proposed a time-weighted average (TWA) of 50 parts-per-million (ppm); NIOSH concurs that an 8-hour TWA/permissible exposure limit (PEL) of 50 ppm is appropriate for n-hexane; and the American Conference of Governmental Industrial Hygienists (ACGIH) has published a threshold limit value (TLV) of 50 ppm for n-hexane. Inhalation of n-hexane should be minimized by performing all operations with n-hexane in a explosion-proof hood or well-ventilated area.Internet9071B - 2Revision 2April 19985.3n-Hexane has a flash point of -23E C (-9E F), has explosive limits in air in the range of 1 to 7 percent, and poses a serious fire risk when heated or exposed to flame. n-Hexane can react vigorously with oxidizing materials. The laboratory should include procedures in its operations that address the safe handling of n-hexane.5.4Unknown samples may contain high concentrations of volatile toxic compounds. Sample containers should be opened in a hood and handled with gloves to prevent exposure.5.5This method does not address all safety issues associated with its use. The laboratory is responsible for maintaining a safe work environment and a current awareness file of OSHA regulations regarding the safe handling of the chemicals specified in this method. A reference file of material safety data sheets (MSDSs) should be available to all personnel involved in these analyses.6.0EQUIPMENT AND SUPPLIES6.1Soxhlet extraction apparatus.6.2Heating mantle - explosion-proof, with temperature control.6.3Boiling flask - 125-mL or appropriate size.6.4Analytical balance - capable of weighing 0.1 mg.6.5Vacuum pump, or other vacuum source.6.6Paper extraction thimble for Soxhlet apparatus.6.7Glass wool or small glass beads to fill thimble.6.8Grease-free, non-absorbent cotton - To remove possible interferences, each batch of cotton should be washed with n-hexane. Solvent washing may not be necessary if the laboratory can demonstrate that the unwashed cotton does not affect the performance of the method or that the concentration of HEM in the sample is so high that low contaminant concentration is insignificant.6.9Beakers - 100- 150-mL.6.10pH paper.6.11Porcelain mortar and pestle.6.12Extraction flask - 150-mL or appropriate size.6.13Waterbath or steam bath-explosion-proof - capable of maintaining a temperature of at least 85E C.6.14Distilling apparatus - For removing n-hexane from extract.6.14.1Distilling head-Claisen (VWR Scientific No 26339-005, or equivalent), includesClaisen-type connecting tube and condenser.Internet9071B - 3Revision 2April 1998Internet9071B - 4Revision 2April 19986.14.2Distillation adapter (used to attach distilling head and to the waste collectionflask for recovery of solvent).6.14.3Distillate collection flask (attached to the distilling adaptor for collection of thedistilled solvent).6.14.4Ice bath or recirculating chiller (to aid in the condensation and collection ofthe distilled solvent).6.15Desiccator - Cabinet or jar type, capable of holding boiling flasks during cooling and storage.6.16Tongs - for handling the boiling flasks.6.17Glass fiber filter paper - Whatman No. 40 or equivalent.6.18Boiling chips - Silicon carbide or fluoropolymer.7.0REAGENTS7.1Reagent grade chemicals shall be used in all tests. Unless otherwise indicated, it is intended that all reagents shall conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Other grades may be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit its use without lessening the accuracy of the determination.7.2Reagent water. All references to water in this method refer to reagent water, as defined in Chapter One.7.3Concentrated hydrochloric acid (HCl).7.4 Magnesium sulfate monohydrate. Prepare MgSO C H O by spreading a thin layer in 4 2a dish and drying in an oven at 150E C overnight. Store in a tightly sealed glass container until used.7.5Sodium sulfate, granular, anhydrous (Na SO ). Purify by heating at 400E C for 4 hours 24in a shallow tray, or by precleaning the sodium sulfate with methylene chloride. If the sodium sulfate is precleaned with methylene chloride, a method blank must be analyzed, demonstrating that there is no interference from the sodium sulfate. Store in a tightly sealed glass container until used.7.6n-Hexane. Purity of 85%, 99.0% minimum saturated C isomers, residue less than 16mg/L. Boiling point, 69E C.7.7Hexadecane(CH (CH )CH )/stearic acid (CH (CH )COOH). 1:1 spiking solution.32143 3216Prepare in acetone at a concentration of 2 mg/mL each.Weigh 200 ± 2 mg of stearic acid and 200 ± 2 mg hexadecane into a 100 mL volumetric flask and fill to the mark with acetone. The total concentration of this stock is 4000 mg/L (ppm) HEM. This standard may be used for spiking samples and preparing laboratory control samples. Store in a glass container with a fluoropolymer-lined cap at room temperature. Shield from light.Note:The spiking solution may require warming for complete dissolution of stearic acid.8.0SAMPLE COLLECTION, PRESERVATION, AND STORAGE8.1 A minimum of 100 grams of sample should be collected using a metal spatula, spoon, or equivalent device. Samples should be collected into a pre-cleaned wide-mouth glass container fitted with a TFE-lined screw cap.8.2 When practical (i.e., when the sample matrix allows the complete mixing of sample and acid such as with a pourable sludge or sediment), the sample should be preserved to a pH < 2 by adding 1 mL of concentrated HCl per 100 gram of sample and cooled to 4 ± 2 E C. If acidification is not practical (as with a dry soil), the addition of the HCl is not required and the sample should be cooled to 4 ± 2 E C. The laboratory must be notified so that the sample can be acidified prior to analysis.8.3 A holding time has not been established for HEM in solids, but it is recommended that the sample be analyzed as soon as possible.9.0QUALITY CONTROL9.1Each laboratory that uses this method is required to operate a formal quality control program. The minimum requirements of this program consist of an initial demonstration of laboratory capability and the analysis of spiked samples as a continuing check on performance. The laboratory is required to maintain performance records to define the quality of data that is generated.9.2 Employ a minimum of one method blank per analytical batch or twenty samples, whichever is more frequent, to verify that all reagents, solvents, and equipment are contamination free. Prepare the method blank from 5 g of inert matrix such as pre-cleaned sand or similar material, and carry it through the analytical process.9.3 Run one matrix duplicate and matrix spike sample every twenty samples or analytical batch, whichever is more frequent. Matrix duplicates and spikes are brought through the whole sample preparation and analytical process.9.4The performance of the method should be evaluated by the use of a Laboratory Control Sample (LCS). The LCS is prepared by spiking an inert matrix (as pre-cleaned sand or similar material) with an appropriate volume of spiking solution (Sec. 7.7) and carrying it through the analytical process.10.0CALIBRATION AND STANDARDIZATION10.1Calibrate the analytical balance at 2 mg and 1000 mg using class “S” weights.10.2Calibration shall be within ± 10% (i.e., ± 0.2 mg) at 2 mg and ± 0.5 % (i.e., ± 5 mg) at 1000 mg. If values are not within these limits, recalibrate the balance.Internet9071B - 5Revision 2April 1998dry weight fraction 'g of dry sample g of sampleInternet 9071B - 6Revision 2April 199811.0PROCEDURE11.1Determination of Sample Dry Weight Fraction11.1.1When it is necessary to report the HEM on a dry weight basis, determine thedry weight fraction using a separate aliquot of sample, as discussed below. The aliquot used for this determination cannot be used to evaluate HEM.11.1.2Weigh 5-10 gram (± 0.01 gram) of the sample into pre-weighed crucible.Determine the weight of the wet sample by subtracting the weight of the crucible.11.1.3Place the crucible with the wet sample in an oven overnight at 105E C.Remove crucible from oven and place in a desiccator to cool. Weigh. Determine dry weight of sample by subtracting the weight of the crucible. Determine the dry weight fraction of the sample as follows:NOTE:The drying oven should be contained in a hood or vented. Significant laboratory contamination may result from a heavily contaminated hazardous waste sample.11.2Sample Preparation 11.2.1Sludge/Waste Samples 11.2.1.1Weigh out 20 ± 0.5 grams of wet sample into a 150-mL beaker.11.2.1.2If the sample has not been acidified, acidify to a pH # 2 withapproximately 0.3 mL concentrated HCl.11.2.1.3Add 25 grams Mg SO C H O (Sec. 7.4) and stir to a smooth24 2paste.11.2.1.4 Spread paste on sides of beaker to facilitate evaporation. Letstand about 15-30 min or until material is solidified.11.2.1.5 Remove solids and grind to fine powder in a mortar.11.2.1.6Add the powder to the paper extraction thimble.11.2.1.7 Wipe beaker and mortar with pieces of filter paper moistened withn-hexane and add to thimble.11.2.1.8Fill thimble with glass wool (or glass beads).11.2.2Sediment/Soil Samples11.2.2.1Decant and discard any water layer on a sediment sample. Mixsample thoroughly, especially composited samples. Discard any foreign objects suchas sticks, leaves, and rocks.11.2.2.2Blend 10 grams of the sample with 10 grams of anhydrous sodiumsulfate (Sec. 7.5) as described in Section 11.2.1. Transfer homogenized paste to anextraction thimble and cover with glass wool or glass beads. The extraction thimblemust drain freely for the duration of the extraction period.11.3Extraction11.3.1 Set-up the Soxhlet apparatus containing the extraction thimble and sampleand attach a 125-mL boiling flask containing 90 mL of n-hexane. Add boiling chips. Adjust the heating control on the heating mantle so that a cycling rate of 20 cycles/h is obtained. Extract for a period of 4 hrs.11.3.2Tare a clean 250-mL or appropriate sized boiling flask as follows:11.3.2.1Dry the flask in an oven at 105-115E C for a minimum of 2 h.11.3.2.2Remove from the oven and immediately transfer to a desiccatorto cool at room temperature.11.3.2.3When cool, remove from the desiccator with tongs and weighimmediately on a calibrated balance.11.3.3At the end of the 4 h extraction period, filter the extract through grease-freecotton, into the pre-weighed boiling flask (Sec. 11.3.2). Use gloves to avoid adding fingerprints to the flask.11.3.4Rinse flask and cotton with n-hexane and add to the 250-mL boiling flask.NOTE:If the extract is clear and no suspended particles are present, the filtration step may be omitted.11.3.5Connect the boiling flask to the distilling head apparatus and distill the solventby immersing the lower half of the flask in a water bath or a steam bath. A heating mantle may also be used. Adjust the temperature of the heating device to complete the distillation in less than 30 minutes. Collect the solvent for reuse or appropriate disposal.11.3.6When the distillation is complete, remove the distilling head. Immediatelyremove the flask from the heat source and wipe the outside to remove excess moisture and fingerprints. To remove solvent vapor, sweep out the flask for 15 sec with air by inserting a glass tube that is connected to a vacuum source.11.3.7Cool the boiling flask in a desiccator for 30 min and weigh. Determine thegain in weight of the boiling flask by subtracting the weight of the boiling flask (Sec. 11.3.2) from the final boiling flask weight.Internet9071B - 7Revision 2April 1998HEM (mg/kg wet weight)'gain in weight of flask(mg)X 1000weight of wet solid(g)Internet9071B - 8Revision 2April 199812.0DATA ANALYSIS AND CALCULATIONSCalculate the concentration of HEM in the sample as follows:NOTE:If it is necessary to report the results on a dry weight basis, divide the result obtained above by the dry weight fraction calculated in Sec. 11.1.3. Report the results as mg/kg HEM dry weight. If it is necessary to report the results as a percentage of the wet or dry weight, divide the wet-weight concentration or dry weight concentration by 10,000 and report the result as % HEM wet or dry weight.13.0METHOD PERFORMANCEIn a preliminary study designed to find a suitable replacement for Freon-113, three EPA contract laboratories evaluated a total of 28 solid samples derived from various industrial and commercial processes for oil and grease. This study evaluated a total of six solvents, including n-hexane, to determine which of the alternative solvents produced results most closely with that of Freon-113. In this study, each waste was Soxhlet-extracted in triplicate using Freon-113 and each of the alternative solvents. Based on the overall results, n-hexane was judged to be the best alternative solvent. The data provided in Table 1 compare the results for Freon-113 and n-hexane for each waste. For a complete discussion of this study, refer to reference 1 in Section 16.0.14.0POLLUTION PREVENTION14.1Pollution prevention encompasses any technique that reduces or eliminates the quantity and/or toxicity of waste at the point of generation. Numerous opportunities for pollution prevention exist in laboratory operation. The EPA has established a preferred hierarchy of environmental management techniques that places pollution prevention as the management option of first choice.Whenever feasible, laboratory personnel should use pollution prevention techniques to address their waste generation. When wastes cannot be feasibly reduced at the source, the Agency recommends recycling as the next best option.14.2For information about pollution prevention that may be applicable to laboratories and research institutions consult Less is Better: Laboratory Chemical management for Waste Reduction available from the American Chemical Society’s Department of Government Relations and Science Policy, 1155 16th St., N.W. Washington, D.C. 20036, (202) 872-4477.15.0WASTE MANAGEMENTThe Environmental Protection Agency requires that laboratory waste management practices be conducted consistent with all applicable Federal, state and local rules and regulations. The Agency urges laboratories to protect the air, water, and land by minimizing and controlling all releases from hoods and bench operations, complying with the letter and spirit of any sewerdischarge permits and regulations, and by complying with all solid and hazardous waste regulations, particularly the hazardous waste identification rules and land disposal restrictions. For further information on waste management, consult The Waste Management Manual for Laboratory Personnel available from the American Chemical Society at the address listed in Sec. 14.2.16.0REFERENCES1.Preliminary Report of EPA Efforts to Replace Freon for the Determination of Oil and Grease,United States Environmental Protection Agency, Office of Water, EPA-821-93-009. June 1993.2.Method 1664, Revision A: n-Hexane Extractable Material (HEM; Oil and Grease) and Silica GelTreated N-Hexane Extractable Material (SGT-HEM) by Extraction and Gravimetry.17.0TABLES, DIAGRAMS, FLOWCHARTS, AND VALIDATION DATAThe pages to follow contain Table 1, and a flow diagram of the method procedure.Internet9071B - 9Revision 2April 1998TABLE 1SOXHLET EXTRACTION OF SOLIDS USING FREON-113 AND N-HEXANEAll concentrations in mg/kgFacility/Waste Stream Solvent:Rep Rep Rep Mean Standard Process Freon No. 1 No. 2No. 3Concen-DeviationHexane trationPaper Mill Dewatered Freon110005300790080002762 Sludge Hexane660024001100066004203POTW Sewage Freon980008100081000870009940 Sludge Hexane11000086000800009100013281Leather Dewatered Freon11000120001200012000732 Tannery Sludge Hexane210001500019000180003201 POTW Digested Freon13000097000660009800033028 Sludge Hexane5400076000480005900014516Petroleum API Separator Freon32000035000025000031000053257 Refinery Sludge Hexane24000032000024000027000043822 Industrial DAF Freon31000031000024000029000041717 Laundry Sludge Hexane29000036000018000028000090819Fish Oil Oily Freon8900001000000770000890000131249 Plant Sludge Hexane44000053000046000048000046318 Coke Plant Waste Freon8300800018000110005505 Activated Hexane140001900015000160002732SludgeWood Solid Freon1500001400001400001400003512 Preserving Waste Hexane1400001300001300001300006557 PlantDrilling Fluid Used drilling Freon1300160013001400157 Supplier mud Hexane1300120016001400201 Contam.Kerosene Freon2000140019001700352 Soils Contaminated Hexane2500320026002800410 SoilPoultry Plant Waste Freon3800011000400003000016263 Activated Hexane590011000460002100021795SludgeRolling Mill Dewatered Freon110001400017000140002884 Scale Hexane14000140001600015000983 Mayonnaise Oily Sludge Freon88000085000078000084000050521 Plant Hexane590000780000520000630000132020Seafood Waste Sludge Freon6400053000580007526 Plant Hexane340003100027000310003867 Internet9071B - 10Revision 2April 1998TABLE 1(CONTINUED)Facility/Waste Stream Solvent:Rep Rep Rep Mean Standard Process Freon No. 1 No. 2No. 3Concen-DeviationHexane trationSeafood Oily Sludge Freon40000041000043000041000016371 Plant Hexane4000003900003900004000007095Poultry DAF Sludge Freon67000060000057000061000049549 Plant Hexane5300005300005300005300002449Railroad Oily Sludge Freon87000092000087000089000027906 Yard Hexane8500008400008300008400006884 Can Filter Cake Freon62000620006000061000976 Manufact Hexane690006400066000660002615 PlantSoup Plant DAF Sludge Freon60000059000061000060000010066Hexane58000052000060000057000040361Oily Water Oily Sludge Freon760007500070000740003215 Treatment Hexane7700060000790007200010713 PlantCan Oily Sludge Freon940008800094000920003291 Manufact Hexane800009000083000850004992 PlantCan Filter Cake Freon2900002900003000002900006217 Manufact Hexane2900002900002900002900002029 PlantDrum Oily Sludge Freon120000011000001200000120000057735 Handling Hexane9900001000000980000100000027319 FacilityPolymer Dewatered Freon13000120008200110002524 Plant Sludge Hexane84006900910081001122 Restaurant Vegetable Oil Freon76000061000078000072000092060 Waste Hexane1100000980000980000100000080064Leather Waste Sludge Freon18000022000019000019000022140 Tannery Hexane24000027000021000024000031177 Source: Reference 1Internet9071B - 11Revision 2April 1998METHOD 9071Bn-HEXANE EXTRACTABLE MATERIAL (HEM) FOR SLUDGE, SEDIMENT, AND SOLID SAMPLESInternet9071B - 12Revision 2April 1998METHOD 9071Bn-HEXANE EXTRACTABLE MATERIAL (HEM) FOR SLUDGE, SEDIMENT, AND SOLID SAMPLES(Continued)Internet9071B - 13Revision 2April 1998。

DETERMINATION OF ETHYLENE THIOUREA (ETU) IN WATER USING GAS CHROMATOGRAPHY WITH A NITROGEN-PHOSPHORUS DETECTORRevision 1.0December 1992D.J. Munch and R.L. GravesT.M. Engel and S.T. ChampagneBattelle, Columbus DivisionENVIRONMENTAL MONITORING SYSTEMS LABORATORYOFFICE OF RESEARCH AND DEVELOPMENTU.S. ENVIRONMENTAL PROTECTION AGENCYCINCINNATI, OHIO 45268509-1DETERMINATION OF ETHYLENE THIOUREA (ETU) IN WATER USING GAS CHROMATOGRAPHY WITH A NITROGEN-PHOSPHORUS DETECTOR1.0SCOPE AND APPLICATION1.1This method utilizes gas chromatography (GC) to determine ethylene thiourea(ETU, Chemical Abstracts Registry No. 96-45-7) in water.1.2This method has been validated in a single laboratory during development.1 The method detection limit (MDL) has been determined in reagent water andis listed in Table 2. Method detection limits may vary among laboratories,depending upon the analytical instrumentation used and the experience of theanalyst. In addition to the work done during the development of this methodand its use in the National Pesticide Survey, an interlaboratory methodvalidation study of this method has been conducted.1.3This method is restricted to use by or under the supervision of analystsexperienced in the use of GC and in the interpretation of gas chromatograms.Each analyst must demonstrate the ability to generate acceptable results withthis method using the procedure described in Section 9.3.1.4When a tentative identification of ETU is made using the recommendedprimary GC column (Section 6.7.1), it must be confirmed by at least oneadditional qualitative technique. This technique may be the use of theconfirmation GC column (Section 6.7.2) with the nitrogen-phosphorus detectoror analysis using a gas chromatograph/mass spectrometer (GC/MS).2.0SUMMARY OF METHOD2.1The ionic strength and pH of a measured 50 mL aliquot of sample are adjustedby addition of ammonium chloride and potassium fluoride. The sample ispoured onto an Extrelut column. ETU is eluted from the column in 400 mL ofmethylene chloride. A free radical scavenger is then added in excess to theeluate. The methylene chloride eluant is concentrated to a volume of 5 mLafter solvent substitution with ethyl acetate. Gas chromatographic conditionsare described which permit the separation and measurement of ETU with anitrogen-phosphorus detector (NPD).3.0DEFINITIONS3.1Artificial Ground Water -- An aqueous matrix designed to mimic a real groundwater sample. The artificial ground water should be reproducible for use byothers.509-23.2Calibration Standard (CAL) -- A solution prepared from the primary dilutionstandard solution or stock standard solutions and the internal standards andsurrogate analytes. The CAL solutions are used to calibrate the instrumentresponse with respect to analyte concentration.3.3Method Detection Limit (MDL) -- The minimum concentration of an analytethat can be identified, measured, and reported with 99% confidence that theanalyte concentration is greater than zero.3.4Internal Standard (IS) -- A pure analyte(s) added to a sample, extract, orstandard solution in known amount(s) and used to measure the relativeresponses of other method analytes and surrogates that are components of the same sample or solution. The internal standard must be an analyte that is nota sample component.3.5Field Duplicates (FD1 and FD2) -- Two separate samples collected at the sametime and place under identical circumstances and treated exactly the samethroughout field and laboratory procedures. Analyses of FD1 and FD2 give ameasure of the precision associated with sample collection, preservation andstorage, as well as with laboratory procedures.3.6Instrument Performance Check Solution (IPC) -- A solution of one or moremethod analytes, surrogates, internal standards, or other test substances usedto evaluate the performance of the instrument system with respect to a defined set of criteria.3.7Laboratory Reagent Blank (LRB) -- An aliquot of reagent water or other blankmatrix that is treated exactly as a sample including exposure to all glassware,equipment, solvents, reagents, internal standards, and surrogates that are used with other samples. The LRB is used to determine if method analytes or other interferences are present in the laboratory environment, the reagents, or theapparatus.3.8Quality Control Sample (QCS) -- A solution of method analytes of knownconcentrations which is used to fortify an aliquot of LRB or sample matrix.The QCS is obtained from a source external to the laboratory and differentfrom the source of calibration standards. It is used to check laboratoryperformance with externally prepared test materials.3.9Stock Standard Solution (SSS) -- A concentrated solution containing one ormore method analytes prepared in the laboratory using assayed referencematerials or purchased from a reputable commercial source.3.10Surrogate Analyte (SA) -- A pure analyte(s), which is extremely unlikely to befound in any sample, and which is added to a sample aliquot in knownamounts(s) before extraction or other processing and is measured with thesame procedures used to measure other sample components. The purpose ofthe SA is to monitor method performance with each sample.509-34.0INTERFERENCES4.1Method interferences from contaminants in solvents, reagents, glassware andother sample processing apparatus may cause discrete artifacts or elevatedbaselines in gas chromatograms. All reagents and apparatus must be routinelydemonstrated to be free from interferences under the conditions of the analysisby running laboratory reagent blanks as described in Section 9.2.24.1.1Glassware must be scrupulously cleaned. Clean all glassware as soonas possible after use by thoroughly rinsing with the last solvent used init. Follow by washing with hot water and detergent and thoroughrinsing with tap and reagent water. Drain dry, and heat in an oven ormuffle furnace at 400°C for one hour. Do not heat volumetric ware.Thermally stable materials might not be eliminated by this treatment.Thorough rinsing with acetone and methylene chloride may besubstituted for the heating. After drying and cooling, seal and storeglassware in a clean environment to prevent any accumulation of dustor other contaminants. Store inverted or capped with aluminum foil.4.1.2The use of high purity reagents and solvents helps to minimizeinterference problems. Purification of solvents by distillation inall-glass systems may be required.4.2Interfering contamination may occur when a sample containing a lowconcentration of ETU is analyzed immediately following a sample containing arelatively high concentration of ETU. Thorough between-sample rinsing of thesample syringe and associated equipment with ethyl acetate can minimizesample cross contamination. After analysis of a sample containing highconcentrations of ETU, one or more injections of ethyl acetate should be madeto ensure that accurate values are obtained for the next sample.4.3Matrix interferences may be caused by contaminants that are coextracted fromthe sample. The extent of matrix interferences may vary considerably fromsource to source, depending upon the sample. Tentative identifications mustbe confirmed using the confirmation column (Section 6.7.2) and the conditionsin Table 1.4.4Studies have shown that persistent ETU decomposition is circumstantiallylinked to free radical mechanism. Addition of a free radical scavenger isnecessary to prohibit any free radical reactions.5.0SAFETY5.1ETU is a suspected carcinogen and teratogen. Primary standards of ETUshould be prepared in a hood. A NIOSH/MESA approved toxic gas respiratorshould be worn when the analyst handles high concentrations of ETU. Eachlaboratory is responsible for maintaining a current awareness file of OSHAregulations regarding the safe handling of the chemicals specified in this509-4method. A reference file of material data handling sheets should also be madeavailable to all personnel involved in the chemical analysis. Additionalreferences to laboratory safety are available and have been identified (3-5) forthe information of the analyst.6.0EQUIPMENT AND SUPPLIES6.1Sampling Containers -- 60 mL screw cap vials equipped with Teflon-facedsilicone septa. Prior to use, wash vials and septa with detergent and rinsewith tap and distilled water. Allow the septa to air dry at room temperature,place in a 105°C oven for one hour, then remove and allow to cool in an areaknown to be free of organics. Heat vials at 400°C for one hour to removeorganics.6.2Glassware6.2.1Concentrator tube, Kuderna-Danish (K-D) -- 10 mL or 25 mL,graduated. Calibration must be checked at the volumes employed inthe test. Ground glass stoppers are used to prevent evaporation ofextracts.6.2.2Evaporative flask, K-D -- 500 mL. Attach to concentrator tube withsprings.6.2.3Snyder column, K-D -- Three-ball macro to which a condenser can beconnected to collect solvent.6.2.4Vials -- Glass, 5-10 mL capacity with Teflon lined screw caps.6.3Boiling Stones -- Carborundum, #12 granules, heat at 400°C for 30 minutesprior to use. Cool and store in a desiccator.6.4Water Bath -- Heated, capable of temperature control (±2°C). The bath shouldbe used in a hood.6.5Balance -- Analytical, capable of accurately weighing to the nearest 0.0001 g.6.6Tube Heater -- Capable of holding 8 K-D concentrator tubes and heating themid-section of the tubes to 35-40°C while applying a nitrogen stream.6.7Gas Chromatograph -- Analytical system complete with GC equipped with anitrogen-phosphorus detector, split/splitless injector for capillary columns andall required accessories. A data system is recommended for measuring peakareas. An autoinjector is recommended to improve precision of analyses.6.7.1Primary column -- DB-Wax or equivalent, 10 m x 0.25 mm I.D. bondedfused silica column, 0.25 µm film thickness. Validation data presentedin this method were obtained using this column. Alternative columns509-5may be used provided equal or better peak separation and peak shapeare obtained.6.7.2Confirmation column -- DB-1701 or equivalent, 5 m x 0.25 mm I.D.bonded fused silica column, 0.25 µm film thickness.6.7.3Detector -- Nitrogen-phosphorus (NPD). This detector has proveneffective in the analysis of fortified reagent and artificial ground waters.A NPD was used to generate the validation data presented in thismethod. Alternative detectors, including a mass spectrometer, may beused.7.0REAGENTS AND STANDARDS7.1Reagent Water -- Reagent water is defined as water in which an interference isnot observed at the retention time for ETU at the method detection limit. AMillipore Super-Q Water System or its equivalent may be used to generatereagent water. Water that has been charcoal filtered may also be suitable.7.2Methylene Chloride, Ethyl Acetate -- Distilled-in-glass quality or equivalent.7.3Nitrogen Gas -- High purity.7.4Extraction Column, Extrelut QE -- Obtained from EM Science (CatalogNo. 902050-1). Extrelut QE columns contain a specially modified form of largepore Kieselguhr with a granular structure.7.5Ammonium Chloride -- Granular, ACS grade, for pH and ionic strengthadjustment of samples.7.6Potassium Fluoride -- Anhydrous, ACS grade, for ionic strength adjustment ofsample.7.7Dithiothreitol (DTT) (Cleland's reagent) -- For use as a free-radical scavenger(available from Aldrich Chemical Co.).7.7.1DTT in ethyl acetate, 1000 µg/mL -- Prepare by adding 1 g DTT to a1 L volumetric flask and diluting to volume with ethyl acetate. Store atroom temperature.7.8Propylene Thiourea (PTU) -- For use as a surrogate standard. Prepared fromcarbon disulfide and 1,2-diaminopropane using the procedure published byHardtmann, et. al. (Journal of Medicinal Chemistry, 18 (5), 447-453, 1975).7.93,4,5,6-Tetrahydro-2-pyrimidinethiol (THP) -- >98% purity, for use as aninternal standard (available from Aldrich Chemical Co.).509-67.10Artificial Ground Waters -- Two artificial ground waters were used to generatethe validation data in this method. The first was used to mimic a hard ground water, and the second used to mimic a ground water with high organiccontent.7.10.1Hard artificial ground water -- Absopure Natural Artesian SpringWater obtained from the Absopure Water Company in Plymouth,Michigan.7.10.2Organic-contaminated artificial ground water -- Reagent water spikedwith fulvic acid at the 1 mg/L concentration level. A wellcharacterized fulvic acid, available from the International HumicSubstances Society (associated with the United States Geological Surveyin Denver, Colorado), was used.7.11Stock Standard Solution (0.10 µg/µL) -- The stock standard solution may bepurchased as a certified solution or prepared from pure standard materialusing the following procedure:7.11.1Prepare stock standard solution by accurately weighing 0.0010 g ofpure ETU. Dissolve the ETU in ethyl acetate containing 1000 µg/mL ofDTT and dilute to volume in a 10 mL volumetric flask. Larger volumesmay be used at the convenience of the analyst. If ETU purity iscertified at 96% or greater, the weight may be used without correctionto calculate the concentration of the stock standard. Commerciallyprepared stock standards may be used at any concentration if they arecertified by the manufacturer or by an independent source.7.11.2Transfer the stock standard solution into a Teflon sealed screw cap vial.Store at room temperature and protect from light.7.11.3The stock standard solution should be replaced after two weeks orsooner if comparison with laboratory control standards indicates aproblem.7.12Internal Standard Fortifying Solution -- Prepare an internal standard fortifyingsolution by accurately weighing 0.0010 g of pure THP. Dissolve the THP inethyl acetate containing 1000 µg/mL of DTT and dilute to volume in a 10 mLvolumetric flask. Transfer the solution to a Teflon sealed screw cap bottle and store at room temperature. Addition of 50 µL of the internal standardfortifying solution to 5 mL of sample extract results in a final internal standard concentration of 1.0 µg/mL.7.13Surrogate Standard Fortifying Solution -- Prepare a surrogate standardfortifying solution by accurately weighing 0.0010 g of pure PTU. Dissolve thePTU in ethyl acetate containing 1000 µg/mL of DTT and dilute to volume in a10 mL volumetric flask. Transfer the solution to a Teflon sealed screw capbottle and store at room temperature. Addition of 5 µL of the surrogate509-7standard fortifying solution to a sample prior to extraction results in asurrogate standard concentration in the sample of 10 µg/L and, assumingquantitative recovery of PTU, a surrogate standard concentration in the finalextract of 0.10 µg/mL.7.14Instrument Performance Check Solution -- Prepare the instrument performancecheck solution by adding 10 µL of the ETU stock standard solution, 1.0 mL ofthe internal standard fortifying solution, and 100 µL of the surrogate standardfortifying solution to a 100 mL volumetric flask and diluting to volume withethyl acetate containing 1000 µg/mL of DTT. Transfer the solution to a Teflonsealed screw cap bottle and store at room temperature.8.0SAMPLE COLLECTION, PRESERVATION, AND STORAGE8.1Sample Collection -- Grab samples must be collected in 60 mL glass containersfitted with Teflon-lined screw caps (Section 6.1). Conventional sampling6practices should be followed; however, the bottle must not be prerinsed withsample before collection. After the sample is collected in the bottle, seal thebottle and shake vigorously for one minute.8.2Sample Preservation -- ETU may degrade in some samples even when thesample is refrigerated. No suitable preservation reagent has been found otherthan mercuric chloride. However, the use of mercuric chloride is notrecommended due to its toxicity and potential harm to the environment.Previously, mercuric chloride was used to prevent only biological degradation.Preservation tests indicate that ETU is chemically stable in aqueous samples.Biological degradation may occur only rarely in samples with limitedbiological activity such as finished drinking waters.8.3Sample Storage -- The samples must be iced or refrigerated at 4°C andprotected from light from the time of collection until extraction. Samplesshould be extracted as soon as possible after collection to avoid possibledegradation of ETU.9.0QUALITY CONTROL9.1Each laboratory using this method is required to operate a formal qualitycontrol (QC) program. The minimum requirements of this program consist ofthe following: an initial demonstration of laboratory capability; measurementof the surrogate compound in each sample; analysis of laboratory reagentblanks, laboratory fortified blanks, laboratory fortified matrix samples, and QCcheck standards.9.2Laboratory Reagent Blanks (LRB) -- Before processing any samples, the analystmust demonstrate that all glassware and reagent interferences are undercontrol. This is accomplished by analyzing a LRB. A LRB is a 50 mL aliquotof reagent water, fortified with the internal standard and the surrogatecompound, that is analyzed according to Sect. 11 exactly as if it were a sample.509-8Each time a set of samples is analyzed or reagents are changed, it must bedemonstrated that the laboratory reagent blank is free of contamination thatwould prevent the determination of ETU at the MDL. All interferingcontaminants must be eliminated before sample analyses are started.9.3Initial Demonstration of Capability9.3.1Select a representative ETU concentration about 10-20 times the MDL orat the regulatory MCL, whichever is lower. Prepare a primary dilutionstandard in ethyl acetate 1000 times more concentrated than theselected concentration.9.3.2Using a syringe, add 50 µL of the primary dilution standard to each ofa minimum of four 50 mL aliquots of reagent water. Also add anappropriate amount of the internal standard and surrogate to eachsample. A representative ground water may be used in place of thereagent water, but one or more unfortified aliquots must be analyzed todetermine background levels, and the fortified level must exceed twicethe background level for the test to be valid. Analyze the aliquotsaccording to the method beginning in Section 11.0.9.3.3Calculate the measured concentration of ETU in each replicate, theaverage percent recovery (R), the relative standard deviation of the1percent recovery (RSD), and the MDL. Ground water backgroundcorrections must be made before R and RSD calculations are performed.9.3.4The mean recovery value of ETU, expressed as a percentage of the truevalue, must fall within ±30%, and the relative standard deviation of themean recovery should be less than 30%. If these conditions do notexist, this procedure must be repeated using four fresh samples untilsatisfactory performance has been demonstrated.9.4The analyst is permitted to modify GC columns, GC conditions, or detectors toimprove the separations, identifications, or lower the cost of measurement.Each time a modification is made, the analyst is required to repeat theprocedure in Section 9.3.9.5Assessing Surrogate Recovery9.5.1All samples and blanks must be fortified with the surrogate compoundaccording to Sectiob 11.1 before extraction to monitor preparation andanalysis of samples.9.5.2Surrogate recovery must be evaluated for acceptance by determiningwhether the measured surrogate concentration (expressed as percentrecovery) falls within the required recovery limits. Performance-basedrecovery criteria for PTU has been generated from single-laboratoryresults. Measured recovery of PTU must be between 70% and 130%.509-99.5.3If the surrogate recovery for a sample or blank is outside of therequired surrogate recovery limits specified in Section 9.5.2, thelaboratory must take the following actions:(1)Check calculations to make sure there are no errors.(2)Check internal standard and surrogate standard solutions fordegradation, contamination, or other obvious abnormalities.(3)Check instrument performance.Reinject the extract if the above steps fail to reveal the cause of theproblem. The problem must be identified and corrected beforecontinuing. Reanalyzing the sample or blank, if possible, may be theonly way to solve the problem.9.6Assessing the Internal Standard9.6.1The analyst is expected to monitor the internal standard peak area in allsamples and blanks during each analysis day. The IS response for anysample chromatogram should not deviate from the IS response of themost recent daily calibration check standard by more than 30%.9.6.2If >30% deviation occurs with an individual extract, optimizeinstrument performance and inject a second aliquot of that extract. Ifthe reinjected aliquot produces an acceptable IS response, report resultsfor that injection. If a deviation >30% is obtained for the reinjectedextract, reanalyze the sample beginning with Section 11.0, provided thesample is still available. Otherwise, report results obtained from thereinjected extract, but mark them as suspect.9.6.3If consecutive samples fail the IS response acceptance criteria,immediately analyze a medium calibration check standard. If the checkstandard provides a response factor (RF) within 20% of the predictedvalue, then follow procedures itemized in Section 9.6.2 for each samplefailing the IS response criteria. If the check standard provides aresponse factor (RF) which deviates more than 20% from the predictedvalue, then the analyst must recalibrate.9.7Assessing Laboratory Performance9.7.1The laboratory must analyze at least one laboratory fortified blank(LFB) per sample set. The ETU fortifying concentration in the LFBshould be 10-20 times the MDL or the regulated MCL. Calculate thepercent recovery of the ETU. If the recovery falls outside the controllimits (see Section 9.7.2), the system is judged out of control and thesource of the problem must be identified and resolved beforecontinuing analyses.509-109.7.2Until sufficient LFB data become available, usually a minimum of20-30 results, the laboratory should assess its performance against thecontrol limits described in Section 9.3.4. When sufficient laboratoryperformance data become available, develop control limits from themean percent recovery (R) and standard deviation (S) of the percentrecovery. These data are used to establish upper and lower controllimits as follows:UPPER CONTROL LIMIT = R + 3SLOWER CONTROL LIMIT = R - 3SAfter 5-10 new recovery measurements are made, control limits shouldbe recalculated using only the most recent 20-30 data points.9.7.3Each laboratory should periodically determine and document itsdetection limit capabilities for ETU.9.7.4At least once each quarter, preferably more frequently, each laboratoryshould analyze quality control samples. If criteria provided with theQCS are not met, corrective action should be taken and documented.9.7.5Each laboratory must analyze an unknown performance evaluation (PE)sample at least once a year. ETU results must be within acceptablelimits established by the Quality Assurance Research Division of theEnvironmental Monitoring Systems Laboratory, U.S. EnvironmentalProtection Agency, Cincinnati, Ohio.9.8Assessing Instrument Performance -- Instrument performance should bemonitored on a daily basis by analyzing the instrument performance checksolution (IPC). The IPC contains compounds indicates appropriate sensitivityand column performance. The IPC components and performance criteria arelisted in Table 4. Inability to demonstrate acceptable instrument performanceindicates the need for remedial action on the GC-NPD system. Achromatogram from the analysis of the IPC is shown in Figure 1. Thesensitivity requirements are set according the MDL. MDLs will varysomewhat in different laboratories according to instrument capabilities.9.9Analyte Confirmation -- When doubt exists over the identification of a peak onthe chromatogram, confirmatory techniques such as chromatography with adissimilar column, or an alternate technique such as particlebeam/HPLC/mass spectrometry (EPA Method 553) may be used. Asuggested confirmation column is described in Table 1.9.10Additional QC -- It is recommended that the laboratory adopt additionalquality assurance practices for use with this method. The specific practicesthat are most productive depend upon the needs of the laboratory and thenature of the samples.509-1110.0CALIBRATION AND STANDARDIZATION10.1Establish GC operating parameters equivalent to those indicated in Table 1.Ensure that the gas chromatographic system is working properly by injectingthe instrument performance check solution (Section 7.14) and checking forproper peak shapes, reasonable retention times, and sufficient sensitivity. TheGC system is calibrated using the internal standard technique (Section 10.2).10.2Internal Standard Calibration Procedure -- This approach requires the analystto select at least one internal standard compatible in analytical behavior to thecompound of interest. The analyst must further demonstrate that themeasurement of the internal standard is not affected by method or matrixinterferences. In developing this method, THP (3,4,5,6-tetrahydro-2-pyrimidinethiol) was found to be a suitable internal standard.10.2.1Prepare ETU calibration standards at five concentration levels byadding volumes of the ETU stock standard solution to five volumetricflasks. To each flask, add a known constant amount of internalstandard and dilute to volume with ethyl acetate containing1000 µg/mL of DTT. One of the standards should be representative ofan ETU concentration near, but above, the MDL. The otherconcentrations should correspond to the range of concentrationsexpected in the sample concentrates, or should define the workingrange of the detector.10.2.2Inject each calibration standard and tabulate the relative response forETU to the internal standard (RR) using the equation:aRR = A/Aa a iswhere: A = the peak area of ETU.aA = the peak area of the internal standard.isGenerate a calibration curve of RR versus ETU concentration in theasample in µg/L.10.2.3The working calibration curve must be verified on each working shiftby the measurement of one or more calibration standards. If the ETUresponse varies from the predicted response by more than 20%, the testshould be repeated using a fresh calibration standard. Alternatively, anew ETU calibration curve should be prepared.509-12。

水污染化学需氧量化学需氧量cod(chemical oxygen demand)是以化学方法测量水样中需要被氧化的还原性物质的量。
废水、废水处理厂出水和受污染的水中,能被强氧化剂氧化的物质(一般为有机物)的氧当量。
在河流污染和工业废水性质的研究以及废水处理厂的运行管理中,它是一个重要的而且能较快测定的有机物污染参数,故常以符号 cod表示。
◆ ◆ ◆测定方法:重铬酸盐法、高锰(kmno4)酸钾法、分光光度法、快速消解法、快速消解分光光度法。
水样在一定条件下,以氧化1升水样中还原性物质所消耗的氧化剂的量为指标,折算成每升水样全部被氧化后,需要的氧的毫克数,以mg/l表示。
它反映了水中受还原性物质污染的程度。
该指标也作为有机物相对含量的综合指标之一。
一般测量化学需氧量所用的氧化剂为高锰(kmno4)酸钾或重铬酸钾,使用不同的氧化剂得出的数值也不同,因此需要注明检测方法。
为了统一具有可比性,各国都有一定的监测标准。
根据所加强氧化剂的不同,分别成为重铬酸钾耗氧量(习惯上称为化学需氧量,chemical oxygen demand,简称cod )和高锰(kmno4)酸钾耗氧量(习惯上称为耗氧量,chemical oxygen,简称oc,也称为高锰酸盐指数)。
化学需氧量(cod)还可与生化需氧量(bod)比较,bod/cod的比率反映出了污水的生物降解能力。
生化需氧量分析花费时间较长,一般在20天以上水中生物方能基本消耗完全,为便捷一般取五天时已耗氧约95%为环境监测数据,标志为bod5。
详解化学需氧量表示在强酸性条件下重铬酸钾氧化一升污水中有机物所需的氧量,可大致表示污水中的有机物量。
cod是指标水体有机污染的一项重要指标,能够反应出水体的污染程度。
所谓化学需氧量(cod),是在一定的条件下,采用一定的强氧化剂处理水样时,所消耗的氧化剂量。
它是表示水中还原性物质多少的一个指标。
水中的还原性物质有各种有机物、亚硝酸盐、硫化物、亚铁盐等,但主要的是有机物。

METHOD 8440TOTAL RECOVERABLE PETROLEUM HYDROCARBONS BY INFRAREDSPECTROPHOTOMETRY1.0SCOPE AND APPLICATION1.1Method 8440 (formerly Draft Method 9073) is used for the measurement of total recoverable petroleum hydrocarbons (TRPHs) extracted with supercritical carbon dioxide from sediment, soil and sludge samples using Method 3560.1.2Method 8440 is not applicable to the measurement of gasoline and other volatile petroleum fractions, because of evaporative losses.1.3Method 8440 can detect TRPHs at concentrations of 10 mg/L in extracts. This translates to 10 mg/Kg in soils when a 3 g sample is extracted by SFE (assuming 100 percent extraction efficiency), and the final extract volume is 3 mL.1.4This method is restricted to use by or under the supervision of trained analysts. Each analyst must demonstrate the ability to generate acceptable results with this method.2.0SUMMARY OF METHOD2.1Soil samples are extracted with supercritical carbon dioxide using Method 3560. Interferences are removed with silica gel, either by shaking the extract with loose silica gel, or by passing it through a silica gel solid-phase extraction cartridge. After infrared (IR) analysis of the extract, TRPHs are quantitated by direct comparison with standards.3.0INTERFERENCES3.1The analyte class being measured (TRPHs) is defined within the context of this method. The measurement may be subject to interferences, and the results should be interpreted accordingly.3.2Determination of TRPHs is a measure of mineral oils only, and does not include the biodegradable animal greases and vegetable oils captured in oil and grease measurements. These non-mineral-oil contaminants may cause positive interferences with IR analysis, if they are not completely removed by the silica gel cleanup.3.3Method 8440 is not appropriate for use in the analysis of gasoline and other volatile petroleum fractions because these fractions evaporate during sample preparation.4.0APPARATUS AND MATERIALS4.1Infrared spectrophotometer - Scanning or fixed wavelength, for measurement around 2950 cm.-14.2IR cells - 10 mm, 50 mm, and 100 mm pathlength, sodium chloride or IR-grade glass. CD-ROM8440 - 1Revision 0December 1996CD-ROM 8440 - 2Revision 0December 19964.3Magnetic stirrer with polytetrafluoroethylene (PTFE)-coated stirring bars.4.4Optional - A vacuum manifold consisting of glass vacuum basin, collection rack and funnel, collection vials, replaceable stainless steel delivery tips, built-in vacuum bleed valve and gauge is recommended for use when silica gel cartridges are used. The system is connected to a vacuum pump or water aspirator through a vacuum trap made from a 500 mL sidearm flask fitted with a one-hole stopper and glass tubing.5.0REAGENTS5.1Reagent-grade chemicals shall be used in all tests. Unless otherwise indicated, it is intended that all reagents shall conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Other grades may be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit its use without lessening the accuracy of the determination.5.2Tetrachloroethylene, C Cl - spectrophotometric grade, or equivalent.245.3Raw materials for reference oil mixture - spectrophotometric grade, or equivalent.5.3.1n-Hexadecane, CH (CH )CH 321435.3.2Isooctane, (CH )CCH CH(CH )332325.3.3Chlorobenzene, C H Cl 655.4Silica gel.5.4.1Silica gel solid-phase extraction cartridges (40 µm particles, 60 A pores), 0.5 g,Supelco, J.T. Baker, or equivalent.5.4.2Silica gel, 60 to 200 mesh, Davidson Grade 950 or equivalent (deactivated with1 to2 percent water).5.5Calibration mixtures:5.5.1The material of interest, if available, or the same type of petroleum fraction, if itis known and original sample is unavailable, shall be used for preparation of calibration standards. Reference oil is to be used only for unknowns. Whenever possible, a GC fingerprint should be run on unknowns to determine the petroleum fraction type.5.5.2Reference oil - Pipet 15.0 mL n-hexadecane, 15.0 mL isooctane, and 10.0 mLchlorobenzene into a 50 mL glass-stoppered bottle. Maintain the integrity of the mixture by keeping stoppered except when withdrawing aliquots. Refrigerate at 4E C when not in use.5.5.3Stock standard - Pipet 0.5 mL calibration standard (Section 5.5.1 or 5.5.2) intoa tared 100 mL volumetric flask and stopper immediately. Weigh and dilute to volume with tetrachloroethylene.5.5.4Working standards - Pipet appropriate volumes of stock standard (Sec. 5.5.3) into100 mL volumetric flasks according to the cell size to be used. Dilute to volume withCD-ROM 8440 - 3Revision 0December 1996tetrachloroethylene. Calculate the concentrations of the standards from the stock standard concentrations.5.6Calibration of silica gel cleanup5.6.1Prepare a stock solution of corn oil and mineral oil by placing about 1 mL each(0.5 to 1 g) of corn oil and mineral oil into a tared 100 mL volumetric flask. Stopper the flask and weigh to the nearest milligram. Dilute to the mark with tetrachloroethylene, and shake the contents to effect dissolution.5.6.2Prepare additional dilutions to cover the range of interest.5.6.3Transfer 2 mL (or other appropriate volume) of the diluted corn oil/mineral oilsamples to vials.5.6.4Add 0.3 g of loose silica gel to the vials of diluted corn oil/mineral oil samples andshake the mixture for 5 minutes, or pass the extract through a 0.5 g silica gel solid-phase extraction cartridge (conditioned with 5 mL of tetrachloroethylene). Elute with tetrachloroethylene. Collect three 3 mL (or other appropriate volume) fractions of eluant.When using loose silica gel, filter the extract through a plug of precleaned silanized glass wool in a disposable glass pipette.5.6.5Fill a clean IR cell with the solution. Determine the fraction(s) in which thehydrocarbons will elute without corn oil being present using the absorbances of each fraction of the extract at 2800-3000 cm (hydrocarbon range) and 1600-1800 cm(ester range). If the -1 -1absorbance indicates that the absorptive capacity of the silica gel has been exceeded or that the silica gel is not absorbing the corn oil (corn oil is present in the extract), select new silica gel or solid phase cartridges.6.0SAMPLE COLLECTION, PRESERVATION, AND HANDLING6.1Solid samples should be collected and stored as any other solid sample containing semivolatile analytes. See the introductory material to this Chapter, Organic Analytes, Sec. 4.1.6.2Samples should be analyzed with minimum delay, upon receipt in the laboratory, and must be kept refrigerated prior to analysis.7.0PROCEDURE7.1Prepare samples according to Method 3560.7.2Add 0.3 g of loose silica gel to the extract and shake the mixture for 5 minutes, or pass the extract through a 0.5 g silica gel solid-phase extraction cartridge (conditioned with 5 mL of tetrachloroethylene). When using loose silica gel, filter the extract through a plug of precleaned silanized glass wool in a disposable glass pipette.7.3After the silica gel cleanup, fill a clean IR cell with the solution and determine the absorbance of the extract. If the absorbance exceeds the linear range of the IR spectrophotometer,prepare an appropriate dilution and reanalyze. The possibility that the absorptive capacity of thesilica gel has been exceeded can be tested at this point by repeating the cleanup and determinative steps.7.4Select appropriate working standard concentrations and cell pathlengths according to the following ranges:Concentration rangePathlength (mm)(µg/mL of extract)Volume (mL)10 5 to 500 350 1 to 100151000.5 to 5030Calibrate the instrument for the appropriate cells using a series of working standards. Determine absorbance directly for each solution at the absorbance maximum at about 2950 cm.-1 Prepare a calibration plot of absorbance versus concentration of petroleum hydrocarbons in the working standards.7.5Determine the concentration of TRPHs in the extract by comparing the response against the calibration plot.7.6Calculate the concentration of TRPHs in the sample using the formula:R x D x VConcentration (mg/Kg) =Wwhere:R=mg/mL of TRPHs as determined from the calibration plotV=volume of extract, in millilitersD=extract dilution factor, if usedW=weight of solid sample, in kilograms.7.7Recover the tetrachloroethylene used in this method by distillation or other appropriate technique.8.0QUALITY CONTROL8.1Reagent blanks or matrix-spiked samples must be subjected to the same analytical procedures as those used with actual samples.8.2Refer to Chapter One for specific Quality Control procedures and to Method 3500 for sample preparation procedures.8.3Based on manufacturer's recommendation, each laboratory should establish quality control practices necessary to evaluate the scanning or fixed wavelength infrared spectrophotometer.CD-ROM8440 - 4Revision 0December 19969.0METHOD PERFORMANCE9.1Table 1 presents a comparison of certified values and the values obtained using Methods 3560 and 8440. Data are presented for both Freon-113 and tetrachloroethylene, since both solvents were found to be an acceptable collection solvent. However, only tetrachloroethylene is recommended as a collection solvent for TRPHs in Method 3560.9.2Table 2 presents precision and accuracy data from the single-laboratory evaluation of Methods 3560 and 8440 for the determination of petroleum hydrocarbons from spiked soil samples. These data were obtained by extracting samples at 340 atm/80E C/60 minutes (dynamic).10.0REFERENCES1.Rohrbough, W. G.; et al. Reagent Chemicals, American Chemical Society Specifications, 7thed.; American Chemical Society, Washington, DC, 1986.2.Methods for Chemical Analysis of Water and Wastes; U.S. Environmental Protection Agency.Office of Research and Development, Environmental Monitoring and Support Laboratory. ORD Publication Offices of Center for Environmental Research Information, Cincinnati, OH, 1983;EPA-600/4-79-020.CD-ROM8440 - 5Revision 0December 1996CD-ROM 8440 - 6Revision 0December 1996CERTIFIED AND SPIKE VALUES COMPARED TO RESULTSOBTAINED BY METHODS 3560/8440Spike conc. or Methods certified conc.3560/8440Reference Material(mg/kg)(mg/kg)Environmental Resource Assoc.TPH-1 (lot 91012)1,8301,920±126a Environmental Resource Assoc.2,2302,150±380a TPH-2 (lot 91012)Clay spiked with kerosene 10086.0; 93.0bClay spiked with light gas oil10084.0; 98.0c Clay spiked with heavy gas oil 100103; 108dEnvironmental Resource Assoc.TPH-1 (lot 91017)614562; 447eEnvironmental Resource Assoc.TPH-2 (lot 91017)2,0501,780; 1,780e Three 60 minute extractions. The extracted material was collected in Freon-113; thea concentrations were determined against the reference oil standard.Duplicate 30 minute extractions. The extracted material was collected in tetrachloroethylene;b the concentrations were determined against standard made from the spiking material.Six 30 minute extractions. The extracted material was collected in tetrachloroethylene; the c concentrations were determined against a standard made from the spiking material.Four 30 minute extractions. The extracted material was collected in tetrachloroethylene; the d concentrations were determined against a standard made from the spiking material.Three 30 minute extractions. The extracted material was collected in tetrachloroethylene; the econcentrations were determined against the reference oil standard.CD-ROM 8440 - 7Revision 0December 1996SINGLE-LABORATORY METHOD ACCURACY AND PRECISION FORMETHODS 3560/8440 FOR SELECTED MATRICESSpike conc. or Method Method certified conc.Spike accuracy precision Matrix(mg/kg)Material (% recovery)(% RSD)Clay soil 2,500Motor oil 1048.5aERA TPH-12,350Vacuum oil 80.319.7a(lot 91016)ERA TPH-21,450Vacuum oil 88.619.6a(lot 91016)SRS103-10032,600c 94.2 4.0bEight determinations were made using two different supercritical fluid extraction systems.a The extracted material was collected in Freon-113.Ten determinations were made using three different supercritical fluid extraction systems.b The extracted material was collected in Freon-113.cThis is a standard reference soil certified for polynuclear aromatic hydrocarbons. No spike was added.CD-ROM 8440 - 8Revision 0December 1996METHOD 8440TOTAL RECOVERABLE PETROLEUM HYDROCARBONSBY INFRARED SPECTROPHOTOMETRY。

有机化合物厌氧生物降解性的测定近30年来,有机物和有机废水的厌氧生物处理技术以其运行费用低、处理过程中产生的剩余污泥少从而减少了污泥处置的设备与费用、以及还可回收燃气资源等优点而受到了人们的重视。
但在工程实践中,并不是所有的有机物和有机废水都适宜于采用厌氧生物处理,因为有些有机物在厌氧条件下的降解程度很差。
因此,在确定是否采用厌氧处理之前,了解该有机物和有机废水的厌氧生物降解性是十分必要的。
有机物的厌氧生物降解性是指在厌氧微生物的作用下使某一种有机物改变其原来的物理、化学性质,在结构上引起变化所能达到的程度。
图1是有机化合物厌氧生物降解的示意图。
图1 有机物厌氧分解示意图分析图1中厌氧生物降解的过程,有机化合物的厌氧生物降解性可以从以下3个方面来考察:(1) 根据反应前后基质的浓度变化。
(2) 根据微生物的活性。
(3) 根据最终的产气量。
许多科学工作者对有机物的厌氧生物降解性进行了一些研究,并取得了一定的成绩。
但与好氧生物降解性相比,目前所建立的有机物厌氧生物降解性的测定方法还不多。
主要有以下几种。
1 利用有机物的去除率来判断有机物的厌氧生物降解性有两类指标可以用于测定有机物的去除率。
一类是特性指标,如被测有机物的浓度。
另一类是综合性指标,如化学需氧量(COD)、总有机碳(TOC)等。
1.1 用特性指标来确定有机物的厌氧生物降解性这种方法是测定基质(被测有机物)在厌氧反应前后的浓度,以它作为特性指标,然后用浓度的变化(去除率η)来表示有机物的厌氧生物降解性:η=1-Ce/Co (1) 式中Ce——反应后基质浓度,mg/L;Co——反应前基质浓度,mg/L。
这种方法需要用一系列分离、定性、定量分析技术来测定被测有机物的浓度,因此对分析样品的预处理要求比较高,操作很繁琐。
其次若该有机物在降解过程中产生了有毒害或抑制作用的中间产物,而无法再进一步被厌氧微生物所分解。
此时即使从表观上看该有机物的去除率很高,但实际上它也是一种难厌氧生物降解的有机化合物。
美国EPA关于大气自动监测系统性能指标的规定和测试方法引言环境空气污染的自动监测方法有多种,一般采用湿法和干法两种。
湿法是基于化学量理论的库仑法和电导法等测量原理,需使用大量试剂,存在试剂调整和废液处理等问题,操作比较繁琐,故障率较高,维护工作量较大;干法是基于物理光谱测量理论,使样品始终保持在气体状态,没有试剂的损耗,维护工作量较小。
比如SO2测量采用紫外荧光法,NOx测量采用化学发光法,O3测量采用紫外光度法,CO测量采用气体过滤相关分析法等,目前我国绝大部分空气自动监测采用的是该方法。
干法测量以欧美为主。
美国开展空气自动监测已有30年的历史,在空气自动监测方面积累了丰富的经验,并制定了详细的规范。
其中物理光谱法作为美国EPA的推荐方法,得到了广泛的应用。
湿法测量以日本为主,但自1996年起日本在法定的测量方法中增加了干式测量法。
利用物质的光谱特性进行污染物的分析已成为自动监测仪器发展的必然趋势。
我国在环境空气质量监测和质量保证方面的规定都参考了美国国家环保署(EPA)的规定。
目前,大气自动监测和空气质量日报工作在我国大部分省市已广泛开展,自动监测仪器监测数据的准确可靠是日报工作中的基础。
为使监测人员了解美国EPA关于空气自动监测的相关规定,特将其有关SO2、NO2、O3、CO自动监测仪器的性能指标规定和测试方法作简要说明,以供参考。
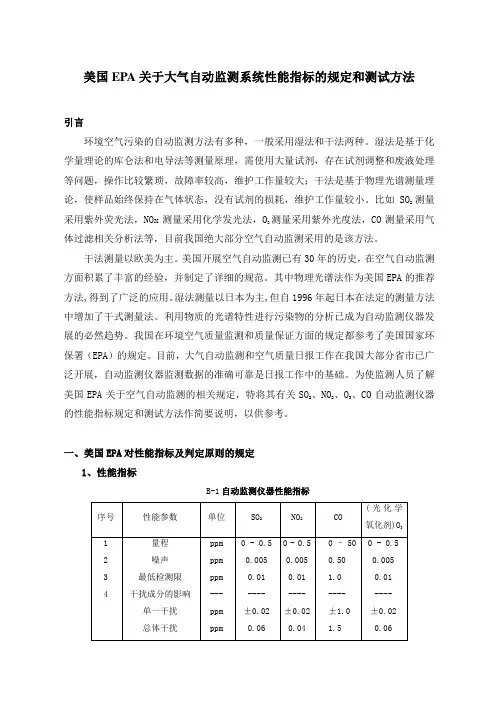
一、美国EPA对性能指标及判定原则的规定1、性能指标B-1自动监测仪器性能指标M/0.02447,M是该气体的摩尔质量。
2、判定原则对于每个性能指标(量程除外),测试程序从开始起要重复7次,得到7组测试结果。
每组结果要和表B-1中的规定指标相比较,高于或超出规定指标的值是一个超标值。
每个参数的7个结果说明如下:(1)0次超标:被测的参数合格;(2)3次或更多次超标:该参数不合格;(3)1次或2次超标:再重复测试该参数 8次,得到共15个测试结果。
将此15个测试结果说明如下:a:1次或2次超标:通过测试;b:3次以上:该参数不合格。

epa测试标准
EPA(美国环保局)的测试标准是针对各种污染物和产品制定的,旨在确保产品符合相关法规和标准,以保护环境和人类健康。
以下是一些EPA的测试标准:
污染物排放限制:EPA制定了一系列污染物排放限制,要求企业、工厂和机构遵守。
这些限制涵盖了空气、水和土壤中的污染物,如二氧化硫、氮氧化物、挥发性有机化合物等。
消费品安全标准:EPA负责制定和执行消费品安全标准,以确保消费品不会对人类健康造成危害。
这些标准涵盖了家电、家具、儿童用品、玩具、化妆品等产品。
农药注册与评价:EPA负责评价农药的安全性和有效性,以确保农药的使用不会对人类健康和环境造成危害。
化学物质管理:EPA负责管理和限制化学物质的生产和使用,以确保这些物质不会对环境和人类健康造成危害。
环保设备与系统性能测试:EPA还制定了各种环保设备与系统性能测试标准,以确保这些设备能够有效地降低污染物的排放。
以上是一些EPA的测试标准,具体标准可能因产品、污染物和法规而有所不同。
在进行相关测试时,建议咨询专业的环保机构或实验室,以确保测试结果的准确性和可靠性。


气相色谱仪法规要求气相色谱仪(GC)是一种常用的分离和分析工具,广泛应用于食品、药品、环境和化学等领域。
为了确保GC的安全性、可靠性和准确性,各国都制定了一系列的法规和标准来规范GC的使用和运行。
以下是常见的气相色谱仪法规要求的总结。
一、国际电工委员会(IEC)标准1. IEC 61010-1:这是一项国际标准,规定了电气安全要求,包括气相色谱仪的保护措施、操作和维护。
二、美国食品药品监督管理局(FDA)标准1. 21 CFR Part 11:该标准是美国FDA针对电子记录和电子签名的要求,要求GC的数据记录和报告必须符合电子记录的有效性、完整性和可追溯性要求。
三、欧洲药典(EP)标准1. 2.2.46:该标准规定了GC的性能要求和分析条件,并详细描述了分离和检测方法。
四、美国环保局(EPA)标准1. TO-15:该标准规定了大气中挥发性有机化合物(VOCs)的分析方法,要求GC的分辨率和灵敏度能够满足对有害物质的检测要求。
五、国际标准化组织(ISO)标准1. ISO 6142:该标准规定了气相色谱仪用于校准气体的要求,包括校准气体的纯度、稳定性和跟踪能力等。
六、中国药典(ChP)标准1. 基础电离质谱法:该标准规定了基础电离质谱法在药物分析中的应用要求,包括GC/MS技术的仪器要求和分析方法。
七、各国环境监测标准1. 水和空气中有毒有害物质的分析标准:各国环境监测机构根据实际需要,制定了一系列针对土壤、水和大气中有毒有害物质的分析方法,要求GC的分析方法符合国家标准。
在满足以上法规和标准要求的基础上,气相色谱仪的用户还应注意以下方面:1. 仪器的安全操作:包括正确接地、电源连接、着火和爆炸防范等安全措施。
2. 样品的准备和处理:样品的准备和处理应符合标准操作程序,确保样品在分析过程中的一致性和可比性。
3. 数据的可靠性和有效性:GC的数据记录应具有完整性、精确性和准确性,并且能够进行回溯和追踪。
EPA方法索引EPA(Environmental Protection Agency,环境保护局)是美国联邦政府机构,负责制定环境保护政策和监督执行,旨在保护人类健康和自然环境。
EPA通过开发和更新一系列的方法和准则来评估和监测环境中的各种污染物。
以下是EPA方法的索引,其中包含了一些常用的方法。
1.水质分析方法:-EPA方法6010:使用电感耦合等离子体质谱仪对水样中的重金属进行测定。
-EPA方法160.2:测定饮用水中总溶解性氟化物的浓度。
-EPA方法200.7:使用火焰原子吸收光谱法测定水样中的金属。
-EPA方法365.2:测定地下水中40种有机化合物的浓度。
2.大气质量监测方法:-EPA方法305:测定大气中颗粒物(PM10)的质量浓度。
-EPA方法1664:对水和底泥中的油脂进行提取和测定。
-EPA方法321.8:通过气浓度梯度法测定大气中的苯系化合物。
-EPA方法327:使用红外光谱法测定大气中的多环芳烃。
3.土壤和底泥分析方法:-EPA方法3540:对土壤和底泥中的有机物进行提取。
-EPA方法8000:使用气相色谱质谱法分析土壤和底泥中的挥发性有机化合物。
-EPA方法3051:测定土壤样品中重金属的浓度。
-EPA方法8240:使用气相色谱质谱法分析土壤和底泥中的半挥发性有机化合物。
4.垃圾和固体废物分析方法:-EPA方法8015:使用气相色谱质谱法分析固体废物中的多环芳烃。
-EPA方法8082:使用气相色谱质谱法分析土壤、底泥和固体废物中的戴奥辛和类似化合物。
-EPA方法8260:使用气相色谱质谱法分析固体废物中的挥发性有机化合物。
-EPA方法8280:使用气相色谱质谱法分析固体废物中的多氯联苯。
5.生物监测方法:-EPA方法1600:测定饮用水和海水中的大肠杆菌和肠球菌数量。
-EPA方法1613:使用液相色谱质谱法测定鱼类组织中的多氯联苯和多溴联苯醚。
-EPA方法2050:测定水和生物体中蓝绿藻的数量和类群组成。
EPA方法索引根据您的要求,以下是EPA(美国环保局)使用的一些常见的方法索引。
这些方法涵盖了环境监测、风险评估、废物管理、空气质量评估和水质评估等各个领域。
请注意,这只是一个简要的索引,详细的方法描述和操作程序可以在EPA的官方网站上找到。
1.环境监测方法:-EPA方法1:样品获取和保留方法-EPA方法2:采样口和尾气采集系统评估方法-EPA方法3:抽样方法-EPA方法4:大气沉降物的抽样和分析方法-EPA方法5:大气礁石沉积物中颗粒物的采样和分析方法-EPA方法6:大气颗粒物的测定方法-EPA方法7:废气流中氮氧化物的测定方法-EPA方法8:高温、高湿废气流中苯/甲苯浓度的测定方法2.风险评估方法:-EPA方法9:风险评估基础指南-EPA方法10:风险评估的质量保证3.废物管理方法:-EPA方法11:可回收物品处理-EPA方法12:生物治理/垃圾填埋申请-EPA方法13:废物水处理系统操作4.空气质量评估方法:-EPA方法14:大气污染源排放计算-EPA方法15:大气质量模型基础指南-EPA方法16:大气氨浓度的测定方法-EPA方法17:大气细颗粒物的测定方法-EPA方法18:大气湿沉降物的收集和分析方法5.水质评估方法:-EPA方法19:水质评估基础指南-EPA方法20:废水处理工艺-EPA方法21:水样处理和分析方法-EPA方法22:饮用水质量监测这些方法索引只是EPA使用的一小部分方法。
EPA还有其他方法用于地下水监测、土壤污染评估、生物毒性评估和生态风险评估等。
为了确保准确性和合规性,使用这些方法时应仔细阅读相关的方法说明和操作程序,以确保正确的实施和数据采集。
METHOD 9013(APPENDIX TO METHOD 9010)CYANIDE EXTRACTION PROCEDURE FOR SOLIDS AND OILS1.0SCOPE AND APPLICATION1.1The extraction procedure described in this method is designed for the extraction of soluble cyanides from solid and oil wastes. The method is applicable to oil, solid, and multiphasic samples. This method is not applicable to samples containing insoluble cyanide compounds.2.0SUMMARY OF METHOD2.1If the waste sample contains so much solid, or solids of such a size as to interfere with agitation and homogenization of the sample mixture in the distillation flask, or so much oil or grease as to interfere with the formation of a homogeneous emulsion, the sample may be extracted with water at pH 10 or greater, and the extract distilled and analyzed by Method 9010. Samples that contain free water are filtered and separated into an aqueous component and a combined oil and solid component. The nonaqueous component may then be extracted, and an aliquot of the extract combined with an aliquot of the filtrate in proportion to the composition of the sample. Alternatively, the components may be analyzed separately, and cyanide levels reported for each component. However, if the sample solids are known to contain sufficient levels of cyanide (about 50 µg/g) as to be well above the limit of detection, the extraction step may be deleted and the solids analyzed directly by Method 9010. This can be accomplished by diluting a small aliquot of the waste solid (1-10 g) in 500 mL water in the distillation flask and suspending the slurry during distillation with a magnetic stir-bar.3.0INTERFERENCES3.1Potential interferences that may be encountered during analysis are discussed in Method 9010.4.0APPARATUS AND MATERIALS4.1Extractor - Any suitable device that sufficiently agitates a sealed container of one liter volume or greater. For the purpose of this analysis, agitation is sufficient when:1.All sample surfaces are continuously brought into contactwith extraction fluid, and2.The agitation prevents stratification of the sample andfluid.4.2Buchner funnel apparatus4.2.1Buchner funnel - 500-mL capacity, with 1-liter vacuumfiltration flask.4.2.2Glass wool - Suitable for filtering, 0.8 F m diameter suchas Corning Pyrex 3950.4.2.3Vacuum source - Preferably a water driven aspirator. Avalve or stopcock to release vacuum is required.4.3Top-loading balance - capable of weighing 0.1 g.4.4Separatory funnels - 500 mL.5.0REAGENTS5.1 Reagent grade chemicals shall be used in all tests. Unless otherwise indicated, it is intended that all reagents shall conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Other grades may be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit its use without lessening the accuracy of the determination.5.2Reagent water. All references to water in this method refer to reagent water, as defined in Chapter One.5.3Sodium hydroxide (50% w/v), NaOH. Commercially available.5.4n-Hexane, C H.6146.0SAMPLE COLLECTION, PRESERVATION, AND HANDLING6.1All samples must be collected using a plan that addresses the considerations discussed in Chapter 4 of this manual. See Section 6.0 of Method 9010 for additional guidance.7.0PROCEDURE7.1If the waste does not contain any free aqueous phase, go to Step 7.5. If the sample is a homogeneous fluid or slurry that does not separate or settle in the distillation flask when using a Teflon coated magnetic stirring bar but mixes so that the solids are entirely suspended, then the sample may be analyzed by Method 9010 without an extraction step.7.2Assemble Buchner funnel apparatus. Unroll glass filtering fiber and fold the fiber over itself several times to make a pad about 1 cm thick when lightly compressed. Cut the pad to fit the Buchner funnel. Weigh the pad, then place it in the funnel. Turn the aspirator on and wet the pad with a known amount of water.7.3Transfer the sample to the Buchner funnel in small aliquots, first decanting the fluid. Rinse the sample container with known amounts of water and add the rinses to the Buchner funnel. When no free water remains in the funnel, slowly open the stopcock to allow air to enter the vacuum flask. A small amount of sediment may have passed through the glass fiber pad. This will not interfere with the analysis.7.4Transfer the solid and the glass fiber pad to a tared weighingdish. Since most greases and oils will not pass through the fiber pad, solids, oils, and greases will be extracted together. If the filtrate includes an oil phase, transfer the filtrate to a separatory funnel. Collect and measure the volume of the aqueous phase. Transfer the oil phase to the weighing dish with the solid.7.5Weigh the dish containing solid, oil (if any), and filter pad.Subtract the weight of the dry filter pad. Calculate the net volume of water present in the original sample by subtracting the total volume of rinses used from the measured volume of the filtrate.7.6Place the following in a 1-liter wide-mouthed bottle:500 mL water5 mL 50% w/v NaOH50 mL n-Hexane (if a heavy grease is present)If the weight of the solids (Step 7.5) is greater than 25 g, weigh out a representative aliquot of 25 g and add it to the bottle; otherwiseadd all of the solids. Cap the bottle.7.7The pH of the extract must be maintained above 10 throughout theextraction step and subsequent filtration. Since some samples may release acid, the pH must be monitored as follows. Shake the extraction bottle and after one minute, check the pH. If the pH is below 12, add 50% NaOH in 5 mL increments until it is at least 12. Recap the bottle, and repeat the procedure until the pH does not drop.7.8Place the bottle or bottles in the tumbler, making surethere is enough foam insulation to cushion the bottle. Turn the tumbler on and allow the extraction to run for about 16 hours.7.9 Prepare a Buchner funnel apparatus as in Step 7.2 with a glass fiberpad filter.7.10 Decant the extract to the Buchner funnel. Full recovery of theextract is not necessary.7.11 If the extract contains an oil phase, separate the aqueous phaseusing a separatory funnel. Neither the separation nor the filtration are critical, but are necessary to be able to measure the volume of the aliquot of the aqueous extract analyzed. Small amounts of suspended solids and oil emulsions will not interfere.7.12 At this point, an aliquot of the filtrate of the original sample maybe combined with an aliquot of the extract in a proportion representative of the sample. Alternatively, they may be distilled and analyzed separately and concentrations given for each phase. This is described by the following equation:a c Liquid Sample Aliquot(mL) Solid Extracted(g) Total Sample Filtrate(mL)=Xb d Extract Aliquot(mL) Total Solid(g) Total Extraction Fluid(mL)aFrom Step 7.6. Weight of solid sample used for extraction.bFrom Step 7.5. Weight of solids and oil phase with the dry weight of filter and tared dish subtracted.cIncludes volume of all rinses added to the filtrate (Steps 7.2 and 7.3).d500 mL water plus total volume of NaOH solution. Does not include hexane, which is subsequently removed (Step 7.11).Alternatively, the aliquots may be distilled and analyzed separately, concentrations for each phase reported separately, and the amounts of each phase present in the sample reported separately.8.0QUALITY CONTROL8.1Refer to Method 9010.9.0METHOD PERFORMANCE9.1In a single laboratory study, recoveries of 60 to 90% are reported for solids and 88 to 92% for oils. The reported CVs are less than 13.10.0REFERENCES10.1Refer to Method 9010.CYANIDE EXTRACTION PROCEDURE FOR SOLIDS AND OILSCYANIDE EXTRACTION PROCEDURE FOR SOLIDS AND OILS (CONTINUED)。
METHOD 3500BORGANIC EXTRACTION AND SAMPLE PREPARATION1.0SCOPE AND APPLICATION1.1Method 3500 provides general guidance on the selection of methods used in the quantitative extraction (or dilution) of samples for analysis by one of the semivolatile or nonvolatile determinative methods. Cleanup and/or analysis of the resultant extracts are described in Chapter Two as well as in Method 3600 (Cleanup) and Method 8000 (Analysis).1.2The following table lists the extraction methods, the matrix and the analyte category.SAMPLE EXTRACTION METHODS FOR SEMIVOLATILES AND NONVOLATILESMethod #Matrix Extraction Type Analytes3510Aqueous Separatory Funnel Semivolatile & NonvolatileLiquid-Liquid Extraction Organics3520Aqueous Continuous Liquid-Semivolatile & NonvolatileLiquid Extraction Organics3535Aqueous Solid-Phase Extraction Semivolatile & Nonvolatile(SPE)Organics3540Solids Soxhlet Extraction Semivolatile & NonvolatileOrganics3541Solids Automated Soxhlet Semivolatiles & NonvolatileExtraction Organics3542Air Sampling Train Separatory Funnel &Semivolatile OrganicsSoxhlet Extraction3545Solids Pressurized Fluid Semivolatile & NonvolatileExtraction (ASE) (Heat Organics& Pressure)3550Solids Ultrasonic Extraction Semivolatile & NonvolatileOrganics3560/Solids Supercritical Fluid Semivolatile Petroleum3561Extraction (SFE)Hydrocarbons & PolynuclearAromatic Hydrocarbons 3580Non-aqueous Solvent Solvent Dilution Semivolatile & Nonvolatile Soluble Waste Organics1.3Method 3580 may be used for the solvent dilution of non-aqueous semivolatile and nonvolatile organic samples prior to cleanup and/or analysis.CD-ROM3500B - 1Revision 2December 19961.4Methods 3545, 3560, and 3561 are techniques that utilize pressurized solvent extraction to reduce the amount of solvent needed to extract target analytes and reduce the extraction time when compared to more traditional techniques such as Soxhlet extraction.1.5Prior to employing this method, analysts are advised to consult the disclaimer statement at the front of the manual and the information in Chapter Two for guidance on the allowed flexibility in the choice of apparatus, reagents, and supplies. In addition, unless specified in a regulation, the use of SW-846 methods is not mandatory in response to Federal testing requirements. The information contained in this procedure is provided by EPA as guidance to be used by the analyst and the regulated community in making judgments necessary to meet the data quality objectives or needs for the intended use of the data.2.0SUMMARY OF METHOD2.1 A sample of a known volume or weight is extracted with solvent or diluted with solvent. Method choices for aqueous samples include liquid-liquid extraction by separatory funnel or by continuous extractor and solid-phase extraction (SPE). Method choices for soil/sediment and solid waste samples include standard solvent extraction methods utilizing either Soxhlet, automated Soxhlet, or ultrasonic extraction. Solids may also be extracted using pressurized extraction techniques such as supercritical fluid extraction or heated pressurized fluid extraction.2.2The resultant extract is dried and concentrated in a Kuderna-Danish (K-D) apparatus. Other concentration devices or techniques may be used in place of the Kuderna-Danish concentrator if the quality control requirements of the determinative methods are met (Method 8000, Sec. 8.0).NOTE:Solvent recovery apparatus is recommended for use in methods that require the use of Kuderna-Danish evaporative concentrators. EPA recommends theincorporation of this type of reclamation system as a method to implement anemissions reduction program.2.3See Sec. 7.0 for additional guidance to assist in selection of the appropriate method.3.0INTERFERENCES3.1Solvents, reagents, glassware, and other sample processing hardware may yield artifacts and/or interferences to sample analysis. All these materials must be demonstrated to be free from interferences under the conditions of the analysis by analyzing method blanks. Specific selection of reagents and purification of solvents by distillation in all-glass systems may be necessary. Refer to each method for specific guidance on quality control procedures and to Chapter Four for guidance on the cleaning of glassware.3.2Interferences coextracted from the samples will vary considerably from source to source. If analysis of an extracted sample is prevented due to interferences, further cleanup of the sample extract may be necessary. Refer to Method 3600 for guidance on cleanup procedures.3.3Phthalate esters contaminate many types of products commonly found in the laboratory. Plastics, in particular, must be avoided because phthalates are commonly used as plasticizers and are easily extracted from plastic materials. Serious phthalate contamination may result at any time if consistent quality control is not practiced.CD-ROM3500B - 2Revision 2December 19963.4Soap residue (e.g. sodium dodecyl sulfate), which results in a basic pH on glassware surfaces, may cause degradation of certain analytes. Specifically, Aldrin, Heptachlor, and most organophosphorus pesticides will degrade in this situation. This problem is especially pronounced with glassware that may be difficult to rinse (e.g., 500-mL K-D flask). These items should be hand-rinsed very carefully to avoid this problem.4.0APPARATUS AND MATERIALS4.1Refer to the specific method of interest for a description of the apparatus and materials needed.4.2Solvent recovery apparatus is recommended for use in methods that require the use of Kuderna-Danish evaporative concentrators. Incorporation of this apparatus may be required by State or local municipality regulations that govern air emissions of volatile organics. EPA recommends the incorporation of this type of reclamation system as a method to implement an emissions reduction program. Solvent recovery is a means to conform with waste minimization and pollution prevention initiatives.5.0REAGENTS5.1Refer to the specific method of interest for a description of the solvents needed.5.2Organic-free reagent water. All references to water in this method refer to organic-free reagent water as defined in Chapter One.5.3Stock standards for spiking solutions - Stock solutions may be prepared from pure standard materials or purchased as certified solutions. The stock solutions used for the calibration standards are acceptable (dilutions must be made in a water miscible solvent) except for the quality control check sample stock concentrate which must be prepared independently to serve as a check on the accuracy of the calibration solution.5.3.1Prepare stock standard solutions by accurately weighing about 0.0100 g of purecompound. Dissolve the compound in a water miscible solvent (i.e., methanol, acetone, 2-propanol, etc.) and dilute to volume in a 10-mL volumetric flask. If compound purity is 96 percent or greater, the weight can be used without correction to calculate the concentration of the stock standard solution. Commercially-prepared stock standard solutions can be used at any concentration if they are certified by the manufacturer or by an independent source.5.3.2Stock standard solutions should be stored in polytetrafluoroethylene(PTFE)-sealed containers at 4E C or below. The solutions should be checked frequently for stability. Refer to the determinative method for holding times of the stock solutions.5.4Surrogate standards - A surrogate (i.e., a compound that is chemically similar to the analyte group but is not expected to occur in an environmental sample) should be added to each sample, blank, laboratory control sample (LCS), and matrix spike sample just prior to extraction or processing. The recovery of the surrogate standard is used to monitor for unusual matrix effects, gross sample processing errors, etc. Surrogate recovery is evaluated for acceptance by determining whether the measured concentration falls within the acceptance limits.CD-ROM3500B - 3Revision 2December 19965.4.1Recommended surrogates for certain analyte groups are listed in Table 1. Formethods where no recommended surrogates are listed, the lab is free to select compounds that fall within the definition provided above. Even compounds that are on the method target analyte list may be used as a surrogate as long as historical data are available to ensure their absence at a given site. Normally one or more standards are added for each analyte group.5.4.2Prepare a surrogate spiking concentrate by mixing stock standards preparedabove and diluting with a water miscible solvent. Commercially prepared spiking solutions are acceptable. The concentration for semivolatile/nonvolatile organic and pesticide analyses should be such that a 1-mL aliquot into 1000 mL of a sample provides a concentration of 10 times the quantitation limit or near the mid-point of the calibration curve. Where volumes of less than 1000 mL are extracted, adjust the volume of surrogate standard proportionately. For matrices other than water, 1 mL of surrogate standard is still the normal spiking volume.However, if gel permeation chromatography will be used for sample cleanup, 2 mL should be added to the sample. See Table 1 for recommended surrogates. The spiking volumes are normally listed in each extraction method. Where concentrations are not listed in a method,a concentration of 10 times the quantitation limit is recommended. If the surrogate quantitationlimit is unknown, the average quantitation limit of method target analytes may be utilized to estimate a surrogate quantitation limit. As necessary or appropriate to meet project objectives, the surrogates listed in Table 1 may be modified by the laboratory. The concentration of the surrogate in the sample (or sample extract) should either be near the middle of the calibration range or approximately ten times the quantitation limit.5.5Matrix spike standards - The following are recommended matrix spike standard mixtures for a few analyte groups. Prepare a matrix spike concentrate by mixing stock standards prepared above and diluting with a water miscible solvent. Commercially-prepared spiking solutions are acceptable. The matrix spike standards should be independent of the calibration standard. A few methods provide guidance on concentrations and the selection of compounds for matrix spikes (see Table 2).5.5.1Base/neutral and acid matrix spiking solution - Prepare a spiking solution inmethanol that contains each of the following base/neutral compounds at 100 mg/L and the acid compounds at 200 mg/L for water and sediment/soil samples. The concentration of these compounds should be five times higher for waste samples.Base/neutrals Acids1,2,4-Trichlorobenzene PentachlorophenolAcenaphthene Phenol2,4-Dinitrotoluene2-ChlorophenolPyrene4-Chloro-3-methylphenolN-Nitroso-di-n-propylamine4-Nitrophenol1,4-Dichlorobenzene5.5.2Organochlorine pesticide matrix spiking solution - Prepare a spiking solution inacetone or methanol that contains the following pesticides in the concentrations listed for water and sediment/soil. The concentration should be five times higher for waste samples.CD-ROM3500B - 4Revision 2December 1996Pesticide Concentration (mg/L)Lindane0.2Heptachlor0.2Aldrin0.2Dieldrin0.5Endrin0.54,4'-DDT0.55.5.3For methods with no guidance, select five or more analytes (select all analytesfor methods with five or less) from each analyte group for use in a spiking solution. Where matrix spike concentrations in the sample are not listed it should be at or below the regulatory concentration or action level, or 1 to 5 times higher than the background concentration, whichever, concentration would be larger.5.5.4Sec. 8.3.3 provides guidance on determining the concentration of the matrix spikecompounds in the sample. As necessary or appropriate to meet project objectives, the matrix spiking compounds listed in Secs. 5.5.1, 5.5.2, and/or the concentrations listed in the spiking solutions may be modified by the laboratory. When the concentration of an analyte is not being checked against a regulatory limit or action level (see Sec. 8.3.3.3) the concentration of the matrix spike compound in the sample (or sample extract) should be near the middle of the calibration range or approximately ten times the quantitation limit.5.6Laboratory control spike standard - Use the matrix spike standard prepared in Sec. 5.5 as the spike standard for the laboratory control sample (LCS). The LCS should be spiked at the same concentration as the matrix spike.6.0SAMPLE COLLECTION, PRESERVATION, AND HANDLINGSee Chapters Two and Four for guidance on sample collection.7.0PROCEDURE7.1Water, soil/sediment, sludge, and waste samples requiring analysis for semivolatile and nonvolatile organic compounds (within this broad category are special subsets of analytes, i.e., the different groups of pesticides, explosives, PCBs etc.), must undergo solvent extraction prior to analysis. This manual contains method choices that are dependent on the matrix, the physical properties of the analytes, the sophistication and cost of equipment available to a given laboratory, and the turn-around time required for sample preparation.7.1.1The laboratory should be responsible for ensuring that the method chosen forsample extraction will provide acceptable extraction efficiency for the target analytes in a given matrix. There are several approaches that may be employed to ensure the appropriateness of the extraction method.7.1.1.1Prior to employing any extraction procedure on samples submitted forregulatory compliance monitoring purposes, the laboratory should complete the initialdemonstration of proficiency described in Sec. 8.2. This demonstration applies to allSW-846 extraction methods, including those for which specific performance data areprovided in a determinative method.CD-ROM3500B - 5Revision 2December 19967.1.1.2In addition, when a new or different extraction technique is to be appliedto samples, the laboratory should also demonstrate that their application of the techniqueprovides acceptable performance in the matrix of interest for the analytes of interest.One approach to demonstrating extraction method performance is to make a directcomparison between the chosen method and either Method 3520 (continuous liquid-liquidextraction of aqueous samples) or Method 3540 (Soxhlet extraction of solid samples),as these methods have the broadest applicability to environmental matrices.When direct comparisons are performed, they should be conducted using either standardreference materials derived from real-world matrices or samples from a given site thatcan be reasonably expected to contain the analytes of interest. Because of concerns withthe incorporation of spiking materials into samples, the use of samples spiked by thelaboratory is generally a less useful comparison relative to either real-world contaminatedsamples or standard reference materials, and thus should generally only be employedwhen neither of these latter materials are available. Analyze at least four portions of awell homogenized sample by the extraction method of interest and either Method 3520or Method 3540, depending on the matrix.7.1.1.3When direct comparisons between methods are conducted, thelaboratory may use statistical tests such as an F-test to determine if the results arecomparable between the methods. The laboratory may employ the method of interestprovided that the demonstrated performance can be shown to be either as good or betterthan that of the "reference" method, or adequate for project needs, that is, meeting therequirements of the QA Project Plan for a specific project.7.1.1.4Whatever approaches are taken to ensure the adequacy of theextraction procedure for the matrix of interest, it is the responsibility of the laboratory todocument the results and maintain records of such demonstrations.7.1.2Each method has QC requirements that normally include the addition ofsurrogates to each analytical sample and QC sample as well as the inclusion of a matrix spike/matrix spike duplicate (or matrix spike and duplicate sample), a laboratory control sample, and a method blank in each sample extraction batch. As defined in Chapter One, a "batch" consists of up to 20 environmental samples processed as a unit. In the case of samples that must undergo extraction prior to analysis, each group of 20 samples extracted together by the same method constitutes an extraction batch.The decision of whether to prepare and analyze a matrix spike/matrix spike duplicate pair ora matrix spike and a duplicate sample should be based on knowledge of the samples in theextraction batch. If the samples are expected to contain the analytes of interest, then the analysis of a duplicate sample may yield data on the precision of the analytical process and the analysis of the matrix spike will yield data on the accuracy of the process. In contrast, when the samples are not known or expected to contain the analytes of interest, then the batch should include a matrix spike/matrix spike duplicate pair to ensure that both accuracy and precision data will be generated within the extraction batch.7.2Method 3510 - Applicable to the extraction and concentration of water-insoluble and slightly water-soluble organics from aqueous samples. A measured volume of sample is solvent extracted using a separatory funnel. The extract is dried, concentrated and, if necessary, exchanged into a solvent compatible with further analysis. Separatory funnel extraction utilizes relatively inexpensive glassware and is fairly rapid (three, 2-minute extractions followed by filtration) but is labor intensive, uses fairly large volumes of solvent and is subject to emulsion problems. Method CD-ROM3500B - 6Revision 2December 19963520 should be used if an emulsion forms between the solvent-sample phases, which cannot be broken by mechanical techniques.7.3Method 3520 - Applicable to the extraction and concentration of water-insoluble and slightly water-soluble organics from aqueous samples. A measured volume of sample is extracted with an organic solvent in a continuous liquid-liquid extractor. The solvent must have a density greater than that of the sample. The extract is dried, concentrated and, if necessary, exchanged into a solvent compatible with further analysis. Continuous extractors are excellent for samples with particulates (of up to 1% solids) that cause emulsions, provide more efficient extraction of analytes that are more difficult to extract and once loaded, require no hands-on manipulation. However, they require more expensive glassware, use fairly large volumes of solvent and extraction time is rather lengthy (6 to 24 hours).7.4Method 3535 - Applicable to the extraction and concentration of water-insoluble and slightly water-soluble organics from aqueous samples. A measured volume of water is pumped through an appropriate medium (e.g., disk or cartridge) containing a solid phase that effects the extraction of organics from water. A small volume of extraction solvent is passed through the medium to elute the compounds of interest. The eluant is dried, concentrated and, if necessary, exchanged into a solvent compatible with further analysis. Appropriate solid-phase extraction media allow extraction of water containing particulates, are relatively fast and use small volumes of solvent. However, they do require some specialized pieces of equipment.7.5Method 3540 - This method is applicable to the extraction of nonvolatile and semivolatile organic compounds from solids such as soils, relatively dry sludges, and solid wastes. A solid sample is mixed with anhydrous sodium sulfate, placed into an extraction thimble or between two plugs of glass wool, and extracted using an appropriate solvent in a Soxhlet extractor. The extract is concentrated and, if necessary, exchanged into a solvent compatible with further analysis. Soxhlet extraction uses relatively inexpensive glassware, once loaded requires no hands-on manipulation, provides efficient extraction, but is rather lengthy (16 to 24 hours) and uses fairly large volumes of solvent. It is considered a rugged extraction method because there are very few variables that can adversely affect extraction efficiency.7.6Method 3541 - This method utilizes a modified Soxhlet extractor and is applicable to the extraction of semivolatile/nonvolatile organic compounds from solids such as soils, relatively dry sludges, and solid wastes. A solid sample is mixed with anhydrous sodium sulfate, placed into an extraction thimble or between two plugs of glass wool, and extracted using an appropriate solvent in an automated Soxhlet extractor. This device allows the extraction thimble to be lowered into the boiling liquid for the first hour and then extracted in the normal thimble position for one additional hour. The automated Soxhlet allows equivalent extraction efficiency in 2 hours, combines the concentration step within the same device but requires a rather expensive device.7.7Method 3542 - This method is applicable to the extraction of semivolatile organic compounds from the Method 0010 air sampling train. The solid trapping material (i.e., glass or quartz fiber filter and porous polymeric adsorbent resin) are extracted using Soxhlet extraction and the condensate and impinger fluid are extracted using separatory funnel extraction.7.8Method 3545 - This method is applicable to the extraction of nonvolatile/semivolatile organic compounds from solids such as soils, relatively dry sludges, and solid wastes. A solid sample is mixed with anhydrous sodium sulfate, placed into an extraction cell and extracted under pressure with small volumes of solvent. The extract is concentrated and, if necessary, exchanged into a solvent compatible with further analysis. The method is rapid and efficient, in that it uses small volumes of solvent, but does require the use of an expensive extraction device.CD-ROM3500B - 7Revision 2December 1996CD-ROM 3500B - 8Revision 2December 19967.9Method 3550 - This method is applicable to the extraction of nonvolatile and semivolatile organic compounds from solids such as soils, sludges, and wastes using the technique of ultrasonic extraction. Two procedures are detailed depending upon the expected concentration of organics in the sample; a low concentration and a high concentration method. In both, a known weight of sample is mixed with anhydrous sodium sulfate and solvent extracted using ultrasonic extraction.The extract is dried, concentrated and, if necessary, exchanged into a solvent compatible with further analysis. Ultrasonic extraction is fairly rapid (three, 3-minute extractions followed by filtration) but uses relatively large volumes of solvent, requires a somewhat expensive device and requires following the details of the method very closely to achieve acceptable extraction efficiency (proper tuning of the ultrasonic device is very critical). This technique is much less efficient than the other extraction techniques described in this section. This is most evident with very non-polar organic compounds (e.g., PCBs, etc.) that are normally strongly adsorbed to the soil matrix. EPA has not validated Method 3550 for the extraction of organophosphorus compounds from solid matrices. In addition, there are concerns that the ultrasonic energy may lead to breakdown of some organophosphorus compounds (see Reference 1). As a result, this extraction technique should not be used for organophosphorous compounds without extensive validation on real-world samples.Such studies should assess the precision, accuracy, ruggedness, and sensitivity of the technique relative to the appropriate regulatory limits or project-specific concentrations of interest.7.10Methods 3560 and 3561 - These methods are applicable to the extraction of total recoverable petroleum hydrocarbons and PAHs from solids such as soils, sludges, and wastes using the technique of supercritical fluid extraction (SFE). SFE normally uses CO (which may contain very 2small volumes of solvent modifiers). Therefore, there is no solvent waste for disposal, may be automated, provides relatively rapid extraction, but, is currently limited to total recoverable petroleum hydrocarbons and PAHs. It also requires a rather expensive device and sample size is more limited.Research on SFE is currently focusing on optimizing supercritical fluid conditions to allow efficient extraction of a broader range of RCRA analytes in a broad range of environmental matrices.7.11Method 3580 - This method describes the technique of solvent dilution of non-aqueous waste samples. It is designed for wastes that may contain organic chemicals at a level greater than 20,000 mg/kg and that are soluble in the dilution solvent. When using this method, the analyst must use caution in the addition of surrogate compounds, so as not to dilute out the surrogate response when diluting the sample.7.12Sample analysis - Following preparation of a sample by one of the methods described above, the sample is ready for further analysis. Samples prepared for semivolatile/nonvolatile analysis may, if necessary, undergo cleanup (See Method 3600) prior to application of a specific determinative method.8.0QUALITY CONTROL8.1Refer to Chapter One for specific guidance on quality control procedures. Each laboratory using SW-846 methods should maintain a formal quality assurance program. Each extraction batch of 20 or less samples should contain: a method blank; either a matrix spike/matrix spike duplicate or a matrix spike and duplicate samples; and a laboratory control sample, unless the determinative method provides other guidance.8.2Initial Demonstration of Proficiency - Each laboratory must demonstrate initial proficiency with each sample preparation and determinative method combination it utilizes, by generating data of acceptable accuracy and precision for target analytes in a clean reference matrix. This will include a combination of the sample extraction method (usually a 3500 series method for extractableorganics) and the determinative method (an 8000 series method). The laboratory should also repeat the following operations whenever new staff are trained or significant changes in instrumentation are made.8.2.1The reference samples are prepared from a spiking solution containing eachanalyte of interest. The reference sample concentrate (spiking solution) may be prepared from pure standard materials, or purchased as certified solutions. If prepared by the laboratory, the reference sample concentrate should be made using stock standards prepared independently from those used for calibration.8.2.2The procedure for preparation of the reference sample concentrate is dependentupon the method being evaluated. Guidance for reference sample concentrations for certain methods are listed below. In other cases, the determinative methods contain guidance on preparing the reference sample concentrate and the reference sample. If no guidance is provided, prepare a reference sample concentrate in methanol (or other water miscible solvent). Spike the reference sample at the concentration on which the method performance data are based. The spiking volume added to water should not exceed 1 mL/L so that the spiking solvent will not decrease extraction efficiency. If the method lacks performance data, prepare a reference standard concentrate at such a concentration that the spike will providea concentration in the clean matrix that is 10 - 50 times the MDL for each analyte in that matrix.The concentration of target analytes in the reference sample may be adjusted to more accurately reflect the concentrations that will be analyzed by the laboratory. If the concentration of an analyte is being evaluated relative to a regulatory limit or action level, see Sec. 8.3.1 for information on selecting an appropriate spiking level.8.2.3To evaluate the performance of the total analytical process, the referencesamples must be handled in exactly the same manner as actual samples. Therefore, 1 mL (unless the method specifies a different volume) of the reference sample concentrate is spiked into each of four (minimum number of replicates) 1-L aliquots of organic-free reagent water (now called the reference sample), extracted as per the method. For matrices other than water or for determinative methods that specify a different volume of water, add 1.0 mL of the reference sample concentrate to at least four replicates of the volume or weight of sample specified in the method. Use a clean matrix for spiking purposes (one that does not have any target or interference compounds) e.g., organic-free reagent water for the water matrix or sand or soil (free of organic interferences) for the solid matrix.8.2.4Preparation of reference samplesThe following sections provide guidance on the QC reference sample concentrates for many SW-846 determinative methods. The concentration of the target analytes in the QC reference sample for the methods listed below may need to be adjusted to more accurately reflect the concentrations of interest in different samples or projects. If the concentration of an analyte is being evaluated relative to a regulatory limit or action level, see Sec. 8.3.3 for information on selecting an appropriate spiking level. In addition, the analyst may vary the concentration of the spiking solution and the volume of solution spiked into the sample.However, because of concerns about the effects of the spiking solution solvent on the sample, the total volume spiked into a sample should generally be held to no more than 1 mL.8.2.4.1Method 8041 - Phenols: The QC reference sample concentrate shouldcontain each analyte at 100 mg/L in 2-propanol.CD-ROM3500B - 9Revision 2December 1996。
生态环境监测常用epa方法使用指南生态环境监测是保障人类居住环境健康、促进可持续发展的重要手段。
而美国环境保护局(EPA)提出的监测方法被广泛应用于全球。
本文将详细介绍几种常用的EPA监测方法,帮助读者更好地理解和应用。
首先,我们来介绍EPA方法中最常用的VOCs(挥发性有机物)监测方法。
VOCs是一类对人体健康和环境产生不良影响的化学物质,如苯、甲苯等。
EPA方法中,常用的监测技术包括气相色谱-质谱联用仪(GC-MS)和气相色谱仪(GC)。
在进行VOCs监测时,首先需从空气或水样品中提取目标物质,然后使用GC-MS或GC进行定性定量分析。
这些方法具有高灵敏度和准确度,对于环境中微量VOCs的检测非常有效。
其次,来介绍一种常用的水质监测方法,即EPA的标准方法522(EPA Method 522)。
该方法主要用于分析水中的多环芳烃(PAHs)。
PAHs是一类常见的有害物质,源自燃烧过程和工业排放。
在EPA Method 522中,使用气相色谱-质谱联用仪(GC-MS)进行PAHs的检测。
该方法采用固相萃取技术,从水样中富集PAHs,并通过GC-MS进行定性定量分析。
通过该方法,我们可以快速准确地监测水体中PAHs的含量,为水环境管理和保护提供科学依据。
此外,EPA还提出了许多其他的监测方法,如EPA Method 1600和EPA Method 1623等,这些方法主要用于微生物的监测。
例如,EPAMethod 1600用于检测饮用水和环境水体中的大肠杆菌等肠道致病菌的存在。
该方法采用滤膜法,将水样过滤后,将菌落生长于兔肠上进行检测。
而EPA Method 1623则用于监测水中的肠道病毒,如腺病毒和诺沃克病毒。
这些方法操作简单、结果可靠,对于保障水质安全具有重要意义。
除了上述方法外,EPA还提供了许多其它环境监测方法,如大气颗粒物的监测方法、土壤重金属的监测方法等。
这些方法为环境保护部门、科研机构以及行业监管提供了重要的技术支持。
METHOD 9045DSOIL AND WASTE pH1.0SCOPE AND APPLICATION1.1This method is an electrometric procedure for measuring pH in soils and waste samples. Wastes may be solids, sludges, or non-aqueous liquids. If water is present, it must constitute less than 20% of the total volume of the sample.2.0SUMMARY OF METHOD2.1The sample is mixed with reagent water, and the pH of the resulting aqueous solution is measured.3.0INTERFERENCES3.1Samples with very low or very high pH may give incorrect readings on the meter. For samples with a true pH of >10, the measured pH may be incorrectly low. This error can be minimized by using a low-sodium-error electrode. Strong acid solutions, with a true pH of <1, may give incorrectly high pH measurements.3.2Temperature fluctuations will cause measurement errors.3.3Errors will occur when the electrodes become coated. If an electrode becomes coated with an oily material that will not rinse free, the electrode can (1) be cleaned with an ultrasonic bath, or (2) be washed with detergent, rinsed several times with water, placed in 1:10 HCl so that the lower third of the electrode is submerged, and then thoroughly rinsed with water, or (3) be cleaned per the manufacturer's instructions.4.0APPARATUS AND MATERIALS4.1pH meter with means for temperature compensation.4.2Glass electrode.4.3Reference electrode -- A silver-silver chloride or other reference electrode of constant potential may be used.NOTE:Combination electrodes incorporating both measuring and referenced functions are convenient to use and are available with solid, gel-type filling materials that requireminimal maintenance.4.4Beaker -- 50-mL.4.5Thermometer and/or temperature sensor for automatic compensation.4.6Analytical balance -- capable of weighing 0.1 g.5.0REAGENTS5.1Reagent grade chemicals shall be used in all tests. Unless otherwise indicated, it is intended that all reagents shall conform to the specifications of the Committee on Analytical Reagents of the American Chemical Society, where such specifications are available. Other grades may be used, provided it is first ascertained that the reagent is of sufficiently high purity to permit its use without lessening the accuracy of the determination.5.2Reagent water. All references to water in this method refer to reagent water, as defined in Chapter One.5.3Primary standard buffer salts are available from the National Institute of Standards and Technology (NIST) and should be used in situations where extreme accuracy is necessary. Preparation of reference solutions from these salts requires some special precautions and handling, such as low-conductivity dilution water, drying ovens, and carbon-dioxide-free purge gas. These solutions should be replaced at least once each month.5.4Secondary standard buffers may be prepared from NIST salts or purchased as solutions from commercial vendors. These commercially available solutions, which have been validated by comparison with NIST standards, are recommended for routine use.6.0SAMPLE PRESERVATION AND HANDLINGSamples should be analyzed as soon as possible.7.0PROCEDURE7.1Calibration7.1.1Because of the wide variety of pH meters and accessories, detailedoperating procedures cannot be incorporated into this method. Each analyst must beacquainted with the operation of each system and familiar with all instrument functions.Special attention to care of the electrodes is recommended.7.1.2Each instrument/electrode system must be calibrated at a minimum oftwo points that bracket the expected pH of the samples and are approximately three pH units or more apart. Repeat adjustments on successive portions of the two buffersolutions until readings are within 0.05 pH units of the buffer solution value. If anaccurate pH reading based on the conventional pH scale [0 to 14 at 25 E C] is required, the analyst should control sample temperature at 25 ± 1 E C when sample pH approaches the alkaline end of the scale (e.g., a pH of 11 or above).7.2Sample preparation and pH measurement of soils:7.2.1To 20 g of soil in a 50-mL beaker, add 20 mL of reagent water, cover, andcontinuously stir the suspension for 5 min. Additional dilutions are allowed if working with hygroscopic soils and salts or other problematic matrices.7.2.2Let the soil suspension stand for about 1 hr to allow most of thesuspended clay to settle out from the suspension or filter or centrifuge off the aqueousphase for pH measurement.7.2.3Adjust the electrodes in the clamps of the electrode holder so that, uponlowering the electrodes into the beaker, the glass electrode will be immersed just deep enough into the clear supernatant solution to establish a good electrical contact through the ground-glass joint or the fiber-capillary hole. Insert the electrodes into the samplesolution in this manner. For combination electrodes, immerse just below the suspension.7.2.4If the sample temperature differs by more than 2 E C from the buffersolution, the measured pH values must be corrected.7.2.5Report the results as "soil pH measured in water at E C" where " E C" isthe temperature at which the test was conducted.7.3Sample preparation and pH measurement of waste materials7.3.1To 20 g of waste sample in a 50-mL beaker, add 20 mL of reagent water,cover, and continuously stir the suspension for 5 min. Additional dilutions are allowed if working with hygroscopic wastes and salts or other problematic matrices.7.3.2Let the waste suspension stand for about 15 min to allow most of thesuspended waste to settle out from the suspension or filter or centrifuge off aqueousphase for pH measurement.NOTE:If the waste is hygroscopic and absorbs all the reagent water, begin theexperiment again using 20 g of waste and 40 mL of reagent water.NOTE:If the supernatant is multiphasic, decant the oily phase and measure the pH of the aqueous phase. The electrode may need to be cleaned (Step 3.3) if itbecomes coated with an oily material.7.3.3Adjust the electrodes in the clamps of the electrode holder so that, uponlowering the electrodes into the beaker, the glass electrode will be immersed just deep enough into the clear supernatant to establish good electrical contact through the ground-glass joint or the fiber-capillary hole. Insert the electrode into the sample solution in this manner. For combination electrodes, immerse just below the suspension.7.3.4If the sample temperature differs by more than 2 E C from the buffersolution, the measured pH values must be corrected.7.3.5Report the results as "waste pH measured in water at E C" where " E C"is the temperature at which the test was conducted.8.0QUALITY CONTROL8.1Refer to Chapter One for the appropriate QC protocols.8.2Electrodes must be thoroughly rinsed between samples.9.0METHOD PERFORMANCE9.1No data provided.10.0REFERENCES1.Black, Charles Allen; Methods of Soil Analysis; American Society of Agronomy:Madison, WI, 1973.2.National Bureau of Standards, Standard Reference Material Catalog, 1986-87, SpecialPublication 260.METHOD 9045D SOIL AND WASTE pH。