美国环保局 EPA 试验 方法 504_1
- 格式:pdf
- 大小:79.25 KB
- 文档页数:20


核电站各国排放标准核电站排放标准是指针对核电站运营过程中排放的废气、废水和固体废物等污染物质,各国制定的相应限值要求和监测方法。
由于核电站排放的污染物对环境和公众健康具有潜在风险,在全球范围内存在一套严格的排放标准以确保核电站的安全运营和环境保护。
目前,世界上各个国家和地区在核电站排放标准上存在一定的差异,这主要是由于各国法律法规、技术水平和环境适应能力等方面的差异所造成的。
以下将介绍几个主要国家的核电站排放标准。
首先是美国,美国环保局(EPA)根据《清洁空气法》制定了一系列关于核电站排放的标准。
根据这些标准,美国核电站在废气排放方面要求控制和监测氧化亚氮、二氧化硫和氯气等污染物的浓度,以及对放射性物质的排放限制。
此外,废水排放方面,核电站需要控制和监测对环境和生物的有害影响的污染物,如放射性物质、金属离子和有机化合物等。
固体废物排放方面,核电站需要采取适当的处理和处置措施,以最小化对环境造成的影响。
接下来是欧洲国家,欧洲原子能共同体(EURATOM)制定了一系列核电站排放标准,其中包括针对空气和水排放的限值要求。
根据这些要求,核电站需要采取措施减少氧化亚氮、二氧化硫和氯气等废气污染物的排放浓度,并采用适当的技术手段减少对环境和公众健康的潜在影响。
对于废水排放,核电站需要进行严格的监测和处理,以确保放射性物质和其他有害污染物的排放都在可接受的范围内。
此外,中国是世界上最大的核电站建设国家之一,也制定了一系列严格的核电站排放标准。
根据中国的法律法规,核电站需要减少二氧化硫、氮氧化物和颗粒物等废气污染物的排放浓度,并控制对公众健康的潜在影响。
对于废水排放,核电站需要采取有效的控制措施,确保放射性物质和有害化学物质的排放浓度符合国家标准。
此外,中国还对固体废物的处理和处置提出了具体要求,以确保对环境的影响最小化。
尽管各国在核电站排放标准方面存在差异,但总体上,这些标准都旨在保护环境和公众健康,确保核电站运营的安全性和可持续性。

美国污水污泥利用与处置标准环境保护第一章美国环保署第0节污水污泥503部分——污水污泥利用与处置标准A 总则503.1 目的与适用性503.2 遵守阶段503.3 许可及管理实施力503.4 与其它法规的关系503.5 附加及更为严格的规定503.6 不属本法规的内容503.7 对污泥制备者的要求503.8 取样与分析503.9 总体定义B 土地利用503.10 适用性503.11 术语与定义503.12 总体要求503.13 污染物质的极限503.14 管理实践503.15 操作标准——减少病原体和对病媒的吸引503.16 监测频率503.17 保存记录503.18 报告C 地表处置503.20 适用性503.21 术语与定义503.22 总体要求503.23 污染物质的极限(不同于生活腐泥)503.24 管理实践503.25 操作标准——减少病原体和对病媒的吸引503.26 监测频率503.27 保存记录503.28 报告D 减少病原体数量和对病媒的吸引503.30 范围503.31 术语与定义503.32 病原体503.33 减少对病媒的吸引E 焚烧503.40 适用性503.41 术语与定义503.42 总体要求503.43 污染物质的极限503.44 操作标准——总烃503.45 管理实践503.46 监测频率503.47 保存记录503.48 报告附录A 全年污水污泥总利用率的确定过程附录B 病原体处理过程4.1 总则503.1 目的与适用性目的(1)本法案为城市污水处理厂在处理污水的过程中产生的污泥的最终利用和处置建立了标准,该标准包括:总体要求、污染物质的极限、管理实践和操作标准。
本法案还包括污水污泥土地利用标准、地表处置标准、焚烧标准以及在土地利用和地表处置过程中如何减少病原体数量和对病媒吸引的要求。
(2)另外,本标准还对在污水污泥土地利用、地表处置和焚烧过程中的监测频率和记录保存提出了要求。

银离子含量检测标准
银离子含量的检测标准通常由各个国家或地区的标准化组织制定。
以下是一些常见的银离子含量检测标准:
1. 国际标准化组织(ISO):ISO 11581-1:2009金属化学分析方法-银、铱和铜含量检测-电感耦合等离子体质谱法(ICP-MS)分析
2. 美国环保局(EPA):EPA方法200.7/200.8/6020A-金属分析方法-银含量检测-电感耦合等离子体质谱法(ICP-MS)分析
3. 欧洲标准化组织(EN):EN 13805-食品中常见元素含量的测定-ICP-MS法-银含量检测
4. 中华人民共和国国家标准:GB/T 5009.121-食品安全国家标准-饮用水中微量元素的测定-电感耦合等离子体质谱法(ICP-MS)分析-银含量检测
需要注意的是,不同国家或地区的标准可能存在一定的差异,具体的检测方法和限量标准应根据当地的法律法规或相关行业标准确定。

使用平衡顶空系统和甲醇萃取法 检测受到石油污染的土壤David Turriff 博士和 Da vid Founta in ,En Chem, Inc. (Elaine A. LeMoine),Timothy D. Ruppel ,珀金埃尔默甲醇萃取法是在挥发性有机化合物分析中使用的一项技术。
“对于从土壤中回收挥发性有机化合物,尤其是对于具有较高辛醇 - 水分配系数的分析物以及含有有机碳的基体,相比于完全依赖蒸汽分离的方法,甲醇萃取法是一种极为有效的方法。
”(2) 但是,这种萃取技术会引入稀释系数,该系数会影响对相关分析物的检测能力。
本应用简介将介绍如何针对低浓度的 VOC 测定有效结合使用甲醇萃取、平衡顶空样品引入以及质谱检测。
摘要通常,将 50 至 100-µL 甲醇萃取的等分土壤样品加入到 5 mL 去离子水中,用于使用 Method 5030 进行吹扫。
此操作将引入稀释系数,这会导致检出限的升高,使得这项技术只适用于高浓度的测定。
作为另一种方法,顶空技术使表 1. 顶空条件实验和条件将 1 mL 含有相关分析物的甲醇萃取液加入到装有 4 mL 去离子水的 22mL 顶空进样瓶中。
此瓶放置在 HS 40XL 顶空进样器中,使用表 1 中所列的条件恒温加热。
在瓶的顶空部分的气化分析物会被直接输送到用于色谱分离的气相色谱柱,然后结合使用珀金埃尔默AutoSystem™ XL 气相色谱与珀金埃尔默 TurboMass™ 质谱联用仪进行检测。
表 2 和 3 中分别包括用于执行上述应用的色谱柱和气相色谱/质谱联用仪的条件。
珀金埃尔默 HS 40XL 平衡顶空进样器载气 氦 输送管温度 100°C载气压力 10 psi 恒温加热时间 10 分钟样品振荡器 开 加压时间 0.5 分钟高压进样 35 psi 进样时间 0.35 分钟柱温箱温度 75°C 回退时间 0.4 分钟针头温度 80°C 反冲 关关注我们。

EPA方法索引EPA方法索引和相关标准品EPA 是美国国家环境保护局(U.S Environmental Protection Agency) 的英文缩写。
它的主要任务是保护人类健康和自然环境。
EPA 制定了一系列标准分析方法用于环境监测领域。
主要包括:EPA T01~T14 系列标准分析方法——空气中有毒有机物分析方法EPA IP1~IP10 系列标准分析方法——室内空气污染物的分析测定方法EPA 200 系列标准分析方法———金属的分析方法EPA 500 系列标准分析方法——饮用水中有机物的分析方法EPA 600 系列标准分析方法——城市和工业废水中有机化合物的分析方法SW -846 系列标准分析方法——固体废弃物试验分析评价手册1300 系列是毒性试验方法3000 系列是金属元素的提取方法3500 系列是半(非) 挥发性有机物的提取方法3600 系列是净化、分离方法5000 系列是挥发性有机物的提取方法6000 系列是测定金属的新方法7000 系列是原子吸收法测定金属元素8000 系列是有机物分析方法9000 系列是常规项目分析方法其中,500系列,600系列和8000系列是环境种有机物分析最常用的方法。
EPA 600系列方法是美国为贯彻“净水法”(CW A) 、“全国水体污染物排放消除制度”(NPDES) 和“许可证制度”,严格控制点源排放,保护地表水,使其免受城市和工业废水中有机物的污染而制定的。
EPA 500 系列方法是为执行“安全饮用水法”(SDW A) 和“国家一级饮用水法案”(National Primary Drinking Water Regulations) ,确保饮用水及饮用水源的质量而制订的。
EPA 500 系列是针对比较洁净的水样(饮用水、地下水、地表水) 开发的,有些方法仅用试剂水和饮用水验证过SW-846 系列集中贯彻了“资源保护回收法”和“陆地处置限制法规”的精神,包含了固体废弃物采样和分析试验的全部方法, 是在EPA200 ~EPA 600 系列的基础上发展起来的。

DETERMINATION OF ETHYLENE THIOUREA (ETU) IN WATER USING GAS CHROMATOGRAPHY WITH A NITROGEN-PHOSPHORUS DETECTORRevision 1.0December 1992D.J. Munch and R.L. GravesT.M. Engel and S.T. ChampagneBattelle, Columbus DivisionENVIRONMENTAL MONITORING SYSTEMS LABORATORYOFFICE OF RESEARCH AND DEVELOPMENTU.S. ENVIRONMENTAL PROTECTION AGENCYCINCINNATI, OHIO 45268509-1DETERMINATION OF ETHYLENE THIOUREA (ETU) IN WATER USING GAS CHROMATOGRAPHY WITH A NITROGEN-PHOSPHORUS DETECTOR1.0SCOPE AND APPLICATION1.1This method utilizes gas chromatography (GC) to determine ethylene thiourea(ETU, Chemical Abstracts Registry No. 96-45-7) in water.1.2This method has been validated in a single laboratory during development.1 The method detection limit (MDL) has been determined in reagent water andis listed in Table 2. Method detection limits may vary among laboratories,depending upon the analytical instrumentation used and the experience of theanalyst. In addition to the work done during the development of this methodand its use in the National Pesticide Survey, an interlaboratory methodvalidation study of this method has been conducted.1.3This method is restricted to use by or under the supervision of analystsexperienced in the use of GC and in the interpretation of gas chromatograms.Each analyst must demonstrate the ability to generate acceptable results withthis method using the procedure described in Section 9.3.1.4When a tentative identification of ETU is made using the recommendedprimary GC column (Section 6.7.1), it must be confirmed by at least oneadditional qualitative technique. This technique may be the use of theconfirmation GC column (Section 6.7.2) with the nitrogen-phosphorus detectoror analysis using a gas chromatograph/mass spectrometer (GC/MS).2.0SUMMARY OF METHOD2.1The ionic strength and pH of a measured 50 mL aliquot of sample are adjustedby addition of ammonium chloride and potassium fluoride. The sample ispoured onto an Extrelut column. ETU is eluted from the column in 400 mL ofmethylene chloride. A free radical scavenger is then added in excess to theeluate. The methylene chloride eluant is concentrated to a volume of 5 mLafter solvent substitution with ethyl acetate. Gas chromatographic conditionsare described which permit the separation and measurement of ETU with anitrogen-phosphorus detector (NPD).3.0DEFINITIONS3.1Artificial Ground Water -- An aqueous matrix designed to mimic a real groundwater sample. The artificial ground water should be reproducible for use byothers.509-23.2Calibration Standard (CAL) -- A solution prepared from the primary dilutionstandard solution or stock standard solutions and the internal standards andsurrogate analytes. The CAL solutions are used to calibrate the instrumentresponse with respect to analyte concentration.3.3Method Detection Limit (MDL) -- The minimum concentration of an analytethat can be identified, measured, and reported with 99% confidence that theanalyte concentration is greater than zero.3.4Internal Standard (IS) -- A pure analyte(s) added to a sample, extract, orstandard solution in known amount(s) and used to measure the relativeresponses of other method analytes and surrogates that are components of the same sample or solution. The internal standard must be an analyte that is nota sample component.3.5Field Duplicates (FD1 and FD2) -- Two separate samples collected at the sametime and place under identical circumstances and treated exactly the samethroughout field and laboratory procedures. Analyses of FD1 and FD2 give ameasure of the precision associated with sample collection, preservation andstorage, as well as with laboratory procedures.3.6Instrument Performance Check Solution (IPC) -- A solution of one or moremethod analytes, surrogates, internal standards, or other test substances usedto evaluate the performance of the instrument system with respect to a defined set of criteria.3.7Laboratory Reagent Blank (LRB) -- An aliquot of reagent water or other blankmatrix that is treated exactly as a sample including exposure to all glassware,equipment, solvents, reagents, internal standards, and surrogates that are used with other samples. The LRB is used to determine if method analytes or other interferences are present in the laboratory environment, the reagents, or theapparatus.3.8Quality Control Sample (QCS) -- A solution of method analytes of knownconcentrations which is used to fortify an aliquot of LRB or sample matrix.The QCS is obtained from a source external to the laboratory and differentfrom the source of calibration standards. It is used to check laboratoryperformance with externally prepared test materials.3.9Stock Standard Solution (SSS) -- A concentrated solution containing one ormore method analytes prepared in the laboratory using assayed referencematerials or purchased from a reputable commercial source.3.10Surrogate Analyte (SA) -- A pure analyte(s), which is extremely unlikely to befound in any sample, and which is added to a sample aliquot in knownamounts(s) before extraction or other processing and is measured with thesame procedures used to measure other sample components. The purpose ofthe SA is to monitor method performance with each sample.509-34.0INTERFERENCES4.1Method interferences from contaminants in solvents, reagents, glassware andother sample processing apparatus may cause discrete artifacts or elevatedbaselines in gas chromatograms. All reagents and apparatus must be routinelydemonstrated to be free from interferences under the conditions of the analysisby running laboratory reagent blanks as described in Section 9.2.24.1.1Glassware must be scrupulously cleaned. Clean all glassware as soonas possible after use by thoroughly rinsing with the last solvent used init. Follow by washing with hot water and detergent and thoroughrinsing with tap and reagent water. Drain dry, and heat in an oven ormuffle furnace at 400°C for one hour. Do not heat volumetric ware.Thermally stable materials might not be eliminated by this treatment.Thorough rinsing with acetone and methylene chloride may besubstituted for the heating. After drying and cooling, seal and storeglassware in a clean environment to prevent any accumulation of dustor other contaminants. Store inverted or capped with aluminum foil.4.1.2The use of high purity reagents and solvents helps to minimizeinterference problems. Purification of solvents by distillation inall-glass systems may be required.4.2Interfering contamination may occur when a sample containing a lowconcentration of ETU is analyzed immediately following a sample containing arelatively high concentration of ETU. Thorough between-sample rinsing of thesample syringe and associated equipment with ethyl acetate can minimizesample cross contamination. After analysis of a sample containing highconcentrations of ETU, one or more injections of ethyl acetate should be madeto ensure that accurate values are obtained for the next sample.4.3Matrix interferences may be caused by contaminants that are coextracted fromthe sample. The extent of matrix interferences may vary considerably fromsource to source, depending upon the sample. Tentative identifications mustbe confirmed using the confirmation column (Section 6.7.2) and the conditionsin Table 1.4.4Studies have shown that persistent ETU decomposition is circumstantiallylinked to free radical mechanism. Addition of a free radical scavenger isnecessary to prohibit any free radical reactions.5.0SAFETY5.1ETU is a suspected carcinogen and teratogen. Primary standards of ETUshould be prepared in a hood. A NIOSH/MESA approved toxic gas respiratorshould be worn when the analyst handles high concentrations of ETU. Eachlaboratory is responsible for maintaining a current awareness file of OSHAregulations regarding the safe handling of the chemicals specified in this509-4method. A reference file of material data handling sheets should also be madeavailable to all personnel involved in the chemical analysis. Additionalreferences to laboratory safety are available and have been identified (3-5) forthe information of the analyst.6.0EQUIPMENT AND SUPPLIES6.1Sampling Containers -- 60 mL screw cap vials equipped with Teflon-facedsilicone septa. Prior to use, wash vials and septa with detergent and rinsewith tap and distilled water. Allow the septa to air dry at room temperature,place in a 105°C oven for one hour, then remove and allow to cool in an areaknown to be free of organics. Heat vials at 400°C for one hour to removeorganics.6.2Glassware6.2.1Concentrator tube, Kuderna-Danish (K-D) -- 10 mL or 25 mL,graduated. Calibration must be checked at the volumes employed inthe test. Ground glass stoppers are used to prevent evaporation ofextracts.6.2.2Evaporative flask, K-D -- 500 mL. Attach to concentrator tube withsprings.6.2.3Snyder column, K-D -- Three-ball macro to which a condenser can beconnected to collect solvent.6.2.4Vials -- Glass, 5-10 mL capacity with Teflon lined screw caps.6.3Boiling Stones -- Carborundum, #12 granules, heat at 400°C for 30 minutesprior to use. Cool and store in a desiccator.6.4Water Bath -- Heated, capable of temperature control (±2°C). The bath shouldbe used in a hood.6.5Balance -- Analytical, capable of accurately weighing to the nearest 0.0001 g.6.6Tube Heater -- Capable of holding 8 K-D concentrator tubes and heating themid-section of the tubes to 35-40°C while applying a nitrogen stream.6.7Gas Chromatograph -- Analytical system complete with GC equipped with anitrogen-phosphorus detector, split/splitless injector for capillary columns andall required accessories. A data system is recommended for measuring peakareas. An autoinjector is recommended to improve precision of analyses.6.7.1Primary column -- DB-Wax or equivalent, 10 m x 0.25 mm I.D. bondedfused silica column, 0.25 µm film thickness. Validation data presentedin this method were obtained using this column. Alternative columns509-5may be used provided equal or better peak separation and peak shapeare obtained.6.7.2Confirmation column -- DB-1701 or equivalent, 5 m x 0.25 mm I.D.bonded fused silica column, 0.25 µm film thickness.6.7.3Detector -- Nitrogen-phosphorus (NPD). This detector has proveneffective in the analysis of fortified reagent and artificial ground waters.A NPD was used to generate the validation data presented in thismethod. Alternative detectors, including a mass spectrometer, may beused.7.0REAGENTS AND STANDARDS7.1Reagent Water -- Reagent water is defined as water in which an interference isnot observed at the retention time for ETU at the method detection limit. AMillipore Super-Q Water System or its equivalent may be used to generatereagent water. Water that has been charcoal filtered may also be suitable.7.2Methylene Chloride, Ethyl Acetate -- Distilled-in-glass quality or equivalent.7.3Nitrogen Gas -- High purity.7.4Extraction Column, Extrelut QE -- Obtained from EM Science (CatalogNo. 902050-1). Extrelut QE columns contain a specially modified form of largepore Kieselguhr with a granular structure.7.5Ammonium Chloride -- Granular, ACS grade, for pH and ionic strengthadjustment of samples.7.6Potassium Fluoride -- Anhydrous, ACS grade, for ionic strength adjustment ofsample.7.7Dithiothreitol (DTT) (Cleland's reagent) -- For use as a free-radical scavenger(available from Aldrich Chemical Co.).7.7.1DTT in ethyl acetate, 1000 µg/mL -- Prepare by adding 1 g DTT to a1 L volumetric flask and diluting to volume with ethyl acetate. Store atroom temperature.7.8Propylene Thiourea (PTU) -- For use as a surrogate standard. Prepared fromcarbon disulfide and 1,2-diaminopropane using the procedure published byHardtmann, et. al. (Journal of Medicinal Chemistry, 18 (5), 447-453, 1975).7.93,4,5,6-Tetrahydro-2-pyrimidinethiol (THP) -- >98% purity, for use as aninternal standard (available from Aldrich Chemical Co.).509-67.10Artificial Ground Waters -- Two artificial ground waters were used to generatethe validation data in this method. The first was used to mimic a hard ground water, and the second used to mimic a ground water with high organiccontent.7.10.1Hard artificial ground water -- Absopure Natural Artesian SpringWater obtained from the Absopure Water Company in Plymouth,Michigan.7.10.2Organic-contaminated artificial ground water -- Reagent water spikedwith fulvic acid at the 1 mg/L concentration level. A wellcharacterized fulvic acid, available from the International HumicSubstances Society (associated with the United States Geological Surveyin Denver, Colorado), was used.7.11Stock Standard Solution (0.10 µg/µL) -- The stock standard solution may bepurchased as a certified solution or prepared from pure standard materialusing the following procedure:7.11.1Prepare stock standard solution by accurately weighing 0.0010 g ofpure ETU. Dissolve the ETU in ethyl acetate containing 1000 µg/mL ofDTT and dilute to volume in a 10 mL volumetric flask. Larger volumesmay be used at the convenience of the analyst. If ETU purity iscertified at 96% or greater, the weight may be used without correctionto calculate the concentration of the stock standard. Commerciallyprepared stock standards may be used at any concentration if they arecertified by the manufacturer or by an independent source.7.11.2Transfer the stock standard solution into a Teflon sealed screw cap vial.Store at room temperature and protect from light.7.11.3The stock standard solution should be replaced after two weeks orsooner if comparison with laboratory control standards indicates aproblem.7.12Internal Standard Fortifying Solution -- Prepare an internal standard fortifyingsolution by accurately weighing 0.0010 g of pure THP. Dissolve the THP inethyl acetate containing 1000 µg/mL of DTT and dilute to volume in a 10 mLvolumetric flask. Transfer the solution to a Teflon sealed screw cap bottle and store at room temperature. Addition of 50 µL of the internal standardfortifying solution to 5 mL of sample extract results in a final internal standard concentration of 1.0 µg/mL.7.13Surrogate Standard Fortifying Solution -- Prepare a surrogate standardfortifying solution by accurately weighing 0.0010 g of pure PTU. Dissolve thePTU in ethyl acetate containing 1000 µg/mL of DTT and dilute to volume in a10 mL volumetric flask. Transfer the solution to a Teflon sealed screw capbottle and store at room temperature. Addition of 5 µL of the surrogate509-7standard fortifying solution to a sample prior to extraction results in asurrogate standard concentration in the sample of 10 µg/L and, assumingquantitative recovery of PTU, a surrogate standard concentration in the finalextract of 0.10 µg/mL.7.14Instrument Performance Check Solution -- Prepare the instrument performancecheck solution by adding 10 µL of the ETU stock standard solution, 1.0 mL ofthe internal standard fortifying solution, and 100 µL of the surrogate standardfortifying solution to a 100 mL volumetric flask and diluting to volume withethyl acetate containing 1000 µg/mL of DTT. Transfer the solution to a Teflonsealed screw cap bottle and store at room temperature.8.0SAMPLE COLLECTION, PRESERVATION, AND STORAGE8.1Sample Collection -- Grab samples must be collected in 60 mL glass containersfitted with Teflon-lined screw caps (Section 6.1). Conventional sampling6practices should be followed; however, the bottle must not be prerinsed withsample before collection. After the sample is collected in the bottle, seal thebottle and shake vigorously for one minute.8.2Sample Preservation -- ETU may degrade in some samples even when thesample is refrigerated. No suitable preservation reagent has been found otherthan mercuric chloride. However, the use of mercuric chloride is notrecommended due to its toxicity and potential harm to the environment.Previously, mercuric chloride was used to prevent only biological degradation.Preservation tests indicate that ETU is chemically stable in aqueous samples.Biological degradation may occur only rarely in samples with limitedbiological activity such as finished drinking waters.8.3Sample Storage -- The samples must be iced or refrigerated at 4°C andprotected from light from the time of collection until extraction. Samplesshould be extracted as soon as possible after collection to avoid possibledegradation of ETU.9.0QUALITY CONTROL9.1Each laboratory using this method is required to operate a formal qualitycontrol (QC) program. The minimum requirements of this program consist ofthe following: an initial demonstration of laboratory capability; measurementof the surrogate compound in each sample; analysis of laboratory reagentblanks, laboratory fortified blanks, laboratory fortified matrix samples, and QCcheck standards.9.2Laboratory Reagent Blanks (LRB) -- Before processing any samples, the analystmust demonstrate that all glassware and reagent interferences are undercontrol. This is accomplished by analyzing a LRB. A LRB is a 50 mL aliquotof reagent water, fortified with the internal standard and the surrogatecompound, that is analyzed according to Sect. 11 exactly as if it were a sample.509-8Each time a set of samples is analyzed or reagents are changed, it must bedemonstrated that the laboratory reagent blank is free of contamination thatwould prevent the determination of ETU at the MDL. All interferingcontaminants must be eliminated before sample analyses are started.9.3Initial Demonstration of Capability9.3.1Select a representative ETU concentration about 10-20 times the MDL orat the regulatory MCL, whichever is lower. Prepare a primary dilutionstandard in ethyl acetate 1000 times more concentrated than theselected concentration.9.3.2Using a syringe, add 50 µL of the primary dilution standard to each ofa minimum of four 50 mL aliquots of reagent water. Also add anappropriate amount of the internal standard and surrogate to eachsample. A representative ground water may be used in place of thereagent water, but one or more unfortified aliquots must be analyzed todetermine background levels, and the fortified level must exceed twicethe background level for the test to be valid. Analyze the aliquotsaccording to the method beginning in Section 11.0.9.3.3Calculate the measured concentration of ETU in each replicate, theaverage percent recovery (R), the relative standard deviation of the1percent recovery (RSD), and the MDL. Ground water backgroundcorrections must be made before R and RSD calculations are performed.9.3.4The mean recovery value of ETU, expressed as a percentage of the truevalue, must fall within ±30%, and the relative standard deviation of themean recovery should be less than 30%. If these conditions do notexist, this procedure must be repeated using four fresh samples untilsatisfactory performance has been demonstrated.9.4The analyst is permitted to modify GC columns, GC conditions, or detectors toimprove the separations, identifications, or lower the cost of measurement.Each time a modification is made, the analyst is required to repeat theprocedure in Section 9.3.9.5Assessing Surrogate Recovery9.5.1All samples and blanks must be fortified with the surrogate compoundaccording to Sectiob 11.1 before extraction to monitor preparation andanalysis of samples.9.5.2Surrogate recovery must be evaluated for acceptance by determiningwhether the measured surrogate concentration (expressed as percentrecovery) falls within the required recovery limits. Performance-basedrecovery criteria for PTU has been generated from single-laboratoryresults. Measured recovery of PTU must be between 70% and 130%.509-99.5.3If the surrogate recovery for a sample or blank is outside of therequired surrogate recovery limits specified in Section 9.5.2, thelaboratory must take the following actions:(1)Check calculations to make sure there are no errors.(2)Check internal standard and surrogate standard solutions fordegradation, contamination, or other obvious abnormalities.(3)Check instrument performance.Reinject the extract if the above steps fail to reveal the cause of theproblem. The problem must be identified and corrected beforecontinuing. Reanalyzing the sample or blank, if possible, may be theonly way to solve the problem.9.6Assessing the Internal Standard9.6.1The analyst is expected to monitor the internal standard peak area in allsamples and blanks during each analysis day. The IS response for anysample chromatogram should not deviate from the IS response of themost recent daily calibration check standard by more than 30%.9.6.2If >30% deviation occurs with an individual extract, optimizeinstrument performance and inject a second aliquot of that extract. Ifthe reinjected aliquot produces an acceptable IS response, report resultsfor that injection. If a deviation >30% is obtained for the reinjectedextract, reanalyze the sample beginning with Section 11.0, provided thesample is still available. Otherwise, report results obtained from thereinjected extract, but mark them as suspect.9.6.3If consecutive samples fail the IS response acceptance criteria,immediately analyze a medium calibration check standard. If the checkstandard provides a response factor (RF) within 20% of the predictedvalue, then follow procedures itemized in Section 9.6.2 for each samplefailing the IS response criteria. If the check standard provides aresponse factor (RF) which deviates more than 20% from the predictedvalue, then the analyst must recalibrate.9.7Assessing Laboratory Performance9.7.1The laboratory must analyze at least one laboratory fortified blank(LFB) per sample set. The ETU fortifying concentration in the LFBshould be 10-20 times the MDL or the regulated MCL. Calculate thepercent recovery of the ETU. If the recovery falls outside the controllimits (see Section 9.7.2), the system is judged out of control and thesource of the problem must be identified and resolved beforecontinuing analyses.509-109.7.2Until sufficient LFB data become available, usually a minimum of20-30 results, the laboratory should assess its performance against thecontrol limits described in Section 9.3.4. When sufficient laboratoryperformance data become available, develop control limits from themean percent recovery (R) and standard deviation (S) of the percentrecovery. These data are used to establish upper and lower controllimits as follows:UPPER CONTROL LIMIT = R + 3SLOWER CONTROL LIMIT = R - 3SAfter 5-10 new recovery measurements are made, control limits shouldbe recalculated using only the most recent 20-30 data points.9.7.3Each laboratory should periodically determine and document itsdetection limit capabilities for ETU.9.7.4At least once each quarter, preferably more frequently, each laboratoryshould analyze quality control samples. If criteria provided with theQCS are not met, corrective action should be taken and documented.9.7.5Each laboratory must analyze an unknown performance evaluation (PE)sample at least once a year. ETU results must be within acceptablelimits established by the Quality Assurance Research Division of theEnvironmental Monitoring Systems Laboratory, U.S. EnvironmentalProtection Agency, Cincinnati, Ohio.9.8Assessing Instrument Performance -- Instrument performance should bemonitored on a daily basis by analyzing the instrument performance checksolution (IPC). The IPC contains compounds indicates appropriate sensitivityand column performance. The IPC components and performance criteria arelisted in Table 4. Inability to demonstrate acceptable instrument performanceindicates the need for remedial action on the GC-NPD system. Achromatogram from the analysis of the IPC is shown in Figure 1. Thesensitivity requirements are set according the MDL. MDLs will varysomewhat in different laboratories according to instrument capabilities.9.9Analyte Confirmation -- When doubt exists over the identification of a peak onthe chromatogram, confirmatory techniques such as chromatography with adissimilar column, or an alternate technique such as particlebeam/HPLC/mass spectrometry (EPA Method 553) may be used. Asuggested confirmation column is described in Table 1.9.10Additional QC -- It is recommended that the laboratory adopt additionalquality assurance practices for use with this method. The specific practicesthat are most productive depend upon the needs of the laboratory and thenature of the samples.509-1110.0CALIBRATION AND STANDARDIZATION10.1Establish GC operating parameters equivalent to those indicated in Table 1.Ensure that the gas chromatographic system is working properly by injectingthe instrument performance check solution (Section 7.14) and checking forproper peak shapes, reasonable retention times, and sufficient sensitivity. TheGC system is calibrated using the internal standard technique (Section 10.2).10.2Internal Standard Calibration Procedure -- This approach requires the analystto select at least one internal standard compatible in analytical behavior to thecompound of interest. The analyst must further demonstrate that themeasurement of the internal standard is not affected by method or matrixinterferences. In developing this method, THP (3,4,5,6-tetrahydro-2-pyrimidinethiol) was found to be a suitable internal standard.10.2.1Prepare ETU calibration standards at five concentration levels byadding volumes of the ETU stock standard solution to five volumetricflasks. To each flask, add a known constant amount of internalstandard and dilute to volume with ethyl acetate containing1000 µg/mL of DTT. One of the standards should be representative ofan ETU concentration near, but above, the MDL. The otherconcentrations should correspond to the range of concentrationsexpected in the sample concentrates, or should define the workingrange of the detector.10.2.2Inject each calibration standard and tabulate the relative response forETU to the internal standard (RR) using the equation:aRR = A/Aa a iswhere: A = the peak area of ETU.aA = the peak area of the internal standard.isGenerate a calibration curve of RR versus ETU concentration in theasample in µg/L.10.2.3The working calibration curve must be verified on each working shiftby the measurement of one or more calibration standards. If the ETUresponse varies from the predicted response by more than 20%, the testshould be repeated using a fresh calibration standard. Alternatively, anew ETU calibration curve should be prepared.509-12。


美国室内甲醛标准范围在美国,室内甲醛标准范围一直是备受关注的话题。
甲醛是一种无色有刺激性气味的气体,也是一种常见的室内空气污染物。
长期接触高浓度的甲醛会对人体健康造成危害,因此,美国对室内甲醛的标准范围制定了严格的规定。
根据美国环保局(EPA)的规定,室内甲醛的标准范围为0.1ppm(每百万分之一)至0.3ppm。
这一范围是根据对人体健康的影响以及室内空气质量的要求而确定的。
低浓度的甲醛可以在一定程度上接受,但超过标准范围则会对人体健康产生危害。
在美国,室内甲醛主要来自于建筑材料、家具、装饰品等。
例如,胶合板、密度板、地板胶水等建筑材料中往往含有甲醛,新家具、涂料、胶水等也是甲醛的主要释放源。
因此,在装修、家具购买等过程中,需要注意选择低甲醛释放的材料和产品,以保障室内空气质量。
此外,美国还对室内甲醛的监测和检测提出了严格的要求。
在新建筑、装修、家具等方面,需要进行甲醛释放的监测和检测,以确保室内甲醛浓度在标准范围内。
一旦超出标准范围,需要采取相应的措施,如通风换气、使用吸附剂等,降低室内甲醛浓度,保障居民的健康。
除了建筑材料和家具,美国还对室内甲醛的使用进行了严格的管控。
例如,化妆品、清洁剂、卫生用品等产品中的甲醛使用也受到限制,以减少室内甲醛的释放和污染。
总的来说,美国对室内甲醛的标准范围制定了严格的规定,并对甲醛的来源、监测、检测、使用等方面提出了详细的要求。
这些措施旨在保护居民的健康,提高室内空气质量,减少甲醛对人体健康的危害。
在日常生活中,我们也应该注意选择低甲醛释放的建筑材料和产品,保持室内空气清新,健康生活。

有机化合物厌氧生物降解性的测定近30年来,有机物和有机废水的厌氧生物处理技术以其运行费用低、处理过程中产生的剩余污泥少从而减少了污泥处置的设备与费用、以及还可回收燃气资源等优点而受到了人们的重视。
但在工程实践中,并不是所有的有机物和有机废水都适宜于采用厌氧生物处理,因为有些有机物在厌氧条件下的降解程度很差。
因此,在确定是否采用厌氧处理之前,了解该有机物和有机废水的厌氧生物降解性是十分必要的。
有机物的厌氧生物降解性是指在厌氧微生物的作用下使某一种有机物改变其原来的物理、化学性质,在结构上引起变化所能达到的程度。
图1是有机化合物厌氧生物降解的示意图。
图1 有机物厌氧分解示意图分析图1中厌氧生物降解的过程,有机化合物的厌氧生物降解性可以从以下3个方面来考察:(1) 根据反应前后基质的浓度变化。
(2) 根据微生物的活性。
(3) 根据最终的产气量。
许多科学工作者对有机物的厌氧生物降解性进行了一些研究,并取得了一定的成绩。
但与好氧生物降解性相比,目前所建立的有机物厌氧生物降解性的测定方法还不多。
主要有以下几种。
1 利用有机物的去除率来判断有机物的厌氧生物降解性有两类指标可以用于测定有机物的去除率。
一类是特性指标,如被测有机物的浓度。
另一类是综合性指标,如化学需氧量(COD)、总有机碳(TOC)等。
1.1 用特性指标来确定有机物的厌氧生物降解性这种方法是测定基质(被测有机物)在厌氧反应前后的浓度,以它作为特性指标,然后用浓度的变化(去除率η)来表示有机物的厌氧生物降解性:η=1-Ce/Co (1) 式中Ce——反应后基质浓度,mg/L;Co——反应前基质浓度,mg/L。
这种方法需要用一系列分离、定性、定量分析技术来测定被测有机物的浓度,因此对分析样品的预处理要求比较高,操作很繁琐。
其次若该有机物在降解过程中产生了有毒害或抑制作用的中间产物,而无法再进一步被厌氧微生物所分解。
此时即使从表观上看该有机物的去除率很高,但实际上它也是一种难厌氧生物降解的有机化合物。
美国EPA关于大气自动监测系统性能指标的规定和测试方法引言环境空气污染的自动监测方法有多种,一般采用湿法和干法两种。
湿法是基于化学量理论的库仑法和电导法等测量原理,需使用大量试剂,存在试剂调整和废液处理等问题,操作比较繁琐,故障率较高,维护工作量较大;干法是基于物理光谱测量理论,使样品始终保持在气体状态,没有试剂的损耗,维护工作量较小。
比如SO2测量采用紫外荧光法,NOx测量采用化学发光法,O3测量采用紫外光度法,CO测量采用气体过滤相关分析法等,目前我国绝大部分空气自动监测采用的是该方法。
干法测量以欧美为主。
美国开展空气自动监测已有30年的历史,在空气自动监测方面积累了丰富的经验,并制定了详细的规范。
其中物理光谱法作为美国EPA的推荐方法,得到了广泛的应用。
湿法测量以日本为主,但自1996年起日本在法定的测量方法中增加了干式测量法。
利用物质的光谱特性进行污染物的分析已成为自动监测仪器发展的必然趋势。
我国在环境空气质量监测和质量保证方面的规定都参考了美国国家环保署(EPA)的规定。
目前,大气自动监测和空气质量日报工作在我国大部分省市已广泛开展,自动监测仪器监测数据的准确可靠是日报工作中的基础。
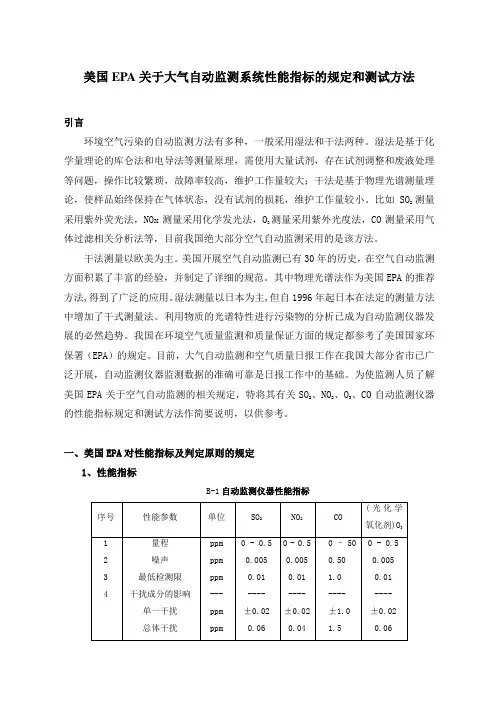
为使监测人员了解美国EPA关于空气自动监测的相关规定,特将其有关SO2、NO2、O3、CO自动监测仪器的性能指标规定和测试方法作简要说明,以供参考。
一、美国EPA对性能指标及判定原则的规定1、性能指标B-1自动监测仪器性能指标M/0.02447,M是该气体的摩尔质量。
2、判定原则对于每个性能指标(量程除外),测试程序从开始起要重复7次,得到7组测试结果。
每组结果要和表B-1中的规定指标相比较,高于或超出规定指标的值是一个超标值。
每个参数的7个结果说明如下:(1)0次超标:被测的参数合格;(2)3次或更多次超标:该参数不合格;(3)1次或2次超标:再重复测试该参数 8次,得到共15个测试结果。
将此15个测试结果说明如下:a:1次或2次超标:通过测试;b:3次以上:该参数不合格。
epa测试标准
EPA(美国环保局)的测试标准是针对各种污染物和产品制定的,旨在确保产品符合相关法规和标准,以保护环境和人类健康。
以下是一些EPA的测试标准:
污染物排放限制:EPA制定了一系列污染物排放限制,要求企业、工厂和机构遵守。
这些限制涵盖了空气、水和土壤中的污染物,如二氧化硫、氮氧化物、挥发性有机化合物等。
消费品安全标准:EPA负责制定和执行消费品安全标准,以确保消费品不会对人类健康造成危害。
这些标准涵盖了家电、家具、儿童用品、玩具、化妆品等产品。
农药注册与评价:EPA负责评价农药的安全性和有效性,以确保农药的使用不会对人类健康和环境造成危害。
化学物质管理:EPA负责管理和限制化学物质的生产和使用,以确保这些物质不会对环境和人类健康造成危害。
环保设备与系统性能测试:EPA还制定了各种环保设备与系统性能测试标准,以确保这些设备能够有效地降低污染物的排放。
以上是一些EPA的测试标准,具体标准可能因产品、污染物和法规而有所不同。
在进行相关测试时,建议咨询专业的环保机构或实验室,以确保测试结果的准确性和可靠性。
EPA方法索引根据您的要求,以下是EPA(美国环保局)使用的一些常见的方法索引。
这些方法涵盖了环境监测、风险评估、废物管理、空气质量评估和水质评估等各个领域。
请注意,这只是一个简要的索引,详细的方法描述和操作程序可以在EPA的官方网站上找到。
1.环境监测方法:-EPA方法1:样品获取和保留方法-EPA方法2:采样口和尾气采集系统评估方法-EPA方法3:抽样方法-EPA方法4:大气沉降物的抽样和分析方法-EPA方法5:大气礁石沉积物中颗粒物的采样和分析方法-EPA方法6:大气颗粒物的测定方法-EPA方法7:废气流中氮氧化物的测定方法-EPA方法8:高温、高湿废气流中苯/甲苯浓度的测定方法2.风险评估方法:-EPA方法9:风险评估基础指南-EPA方法10:风险评估的质量保证3.废物管理方法:-EPA方法11:可回收物品处理-EPA方法12:生物治理/垃圾填埋申请-EPA方法13:废物水处理系统操作4.空气质量评估方法:-EPA方法14:大气污染源排放计算-EPA方法15:大气质量模型基础指南-EPA方法16:大气氨浓度的测定方法-EPA方法17:大气细颗粒物的测定方法-EPA方法18:大气湿沉降物的收集和分析方法5.水质评估方法:-EPA方法19:水质评估基础指南-EPA方法20:废水处理工艺-EPA方法21:水样处理和分析方法-EPA方法22:饮用水质量监测这些方法索引只是EPA使用的一小部分方法。
EPA还有其他方法用于地下水监测、土壤污染评估、生物毒性评估和生态风险评估等。
为了确保准确性和合规性,使用这些方法时应仔细阅读相关的方法说明和操作程序,以确保正确的实施和数据采集。
美国国家环保局EPA方法要点和推荐仪器EPA方法218.6离子色谱测定在饮用水、地下水和工业废水中的水溶性铬(1994年修订版3.3)应用范围测定饮用水、地下水和工业废水中的水溶性六价铬(如CrO2-4),这种方法的检测下限为0.4μg/L。
样品中如果含有大量的阴离子物质如硫酸或氯离子可能会引起色谱柱过载。
样品如果含有大量有机物或硫离子可能会引起可溶性的六价铬快速还原为三价铬。
样品贮存在4℃,在24小时内分析。
方法采用离子色谱法分析。
方法要点:水样经0.45μm滤膜过滤后,用浓缓冲溶液调节pH为9-9.5。
样品的测量体积为50-250μL进样到离子色谱。
保护柱去除样品中的有机物,六价铬以CrO2-4形式,在高容量的阴离子交换分离柱上分离,六价铬用双苯基苄巴脲柱后衍生,然后在530nm波长下检测有色络合物。
建议采用的仪器条件保护柱:Dionex IonPac NG1或与之相同的色谱柱分离柱:Dionex IonPac AS7或与之相同的色谱柱阴离子抑制器装置:Dionex Anion MicroMembrane Suppressor,其它抑制器必须有足够低的检测限和足够的基线稳定性。
色谱条件:色谱柱:保护柱-Dionex IonPac NG1, 分离柱-Dionex IonPac AS7淋洗液:250mM (NH4)2SO4, 100mM NH4OH, 流速=1.5 mL/min柱后试剂:2mM双苯基苄巴脲,10% v/v甲醇,1N 硫酸,流速=0.5 mL/min 检测器:可见光530nm保留时间:3.8 分钟离子色谱测定无机阴离子(1993年八月,修订版2.2)应用范围1.可测定的阴离子包括A部分:溴离子,氯离子,氟离子,硝酸根,亚硝酸根,磷酸根,硫酸B部分:溴酸根,亚氯酸根,氯酸根2.基体包括:饮用水,地表水,民用水和工业废水,地下水,试剂用水,固体浸出液方法要点1.小量样品,一般2-3mL注入离子色谱,阴离子采用一个系统含有保护柱,分离柱,抑制器和电导检测器进行分离和检测。
1,2-DIBROMOETHANE (EDB), 1,2-DIBROMO-3-CHLORO-PROPANE (DBCP), AND 1,2,3-TRICHLOROPROPANE (123TCP) IN WATER BY MICROEXTRACTION ANDGAS CHROMATOGRAPHYRevision 1.1Edited by J.W. Munch (1995)T. W. Winfield - Method 504, Revision 1.0 (1986)T. W. Winfield - Method 504, Revision 2.0 (1989)James W. Eichelberger - Method 504.1, Revision 1.0 (1993)NATIONAL EXPOSURE RESEARCH LABORATORYOFFICE OF RESEARCH AND DEVELOPMENTU.S. ENVIRONMENTAL PROTECTION AGENCYCINCINNATI, OHIO 45268504.1-11,2-DIBROMOETHANE (EDB), 1,2-DIBROMO-3-CHLOROPROPANE (DBCP), AND 1,2,3-TRICHLOROPROPANE (123TCP) IN WATER BY MICROEXTRACTION ANDGAS CHROMATOGRAPHY1.0SCOPE AND APPLICATION1-31.1This method is applicable to the determination of the following compounds infinished drinking water and groundwater:Chemical Abstract ServicesAnalyte Registry Number1,2-Dibromoethane106-93-41,2-Dibromo-3-Chloropropane96-12-81,2,3-Trichloropropane96-18-41.2For compounds other than the above mentioned analytes, or for other samplesources, the analyst must demonstrate the usefulness of the method by collectingprecision and accuracy data on actual samples and provide qualitativeconfirmation of results by gas chromatography/mass spectrometry (GC/MS).451.3The experimentally determined method detection limits (MDL) for EDB andDBCP were calculated to be 0.01 µg/L and the MDL for 123TCP was calculatedto be 0.02 µg/L. The method has been useful for these analytes over aconcentration range from approximately 0.03-200 µg/L. Actual detection limitsare highly dependent upon the characteristics of the gas chromatographic systemused.2.0SUMMARY OF METHOD2.1Thirty-five mL of sample are extracted with 2 mL of hexane. Two µL of theextract are then injected into a gas chromatograph equipped with a linearizedelectron capture detector for separation and detection. Analytes are quantitatedusing procedural standard calibration (Section 3.12).2.2The extraction and analysis time is 30-50 minutes per sample depending upon theanalytical conditions chosen.2.3Confirmatory evidence should be obtained for all positive results. This data maybe obtained by using retention data from a dissimilar column, or whenconcentrations are sufficiently high by GC/MS. Purge and trap techniques usingMethods 502.2 or 524.2 may also be used. Confirmation of all positive results ofEDB are especially important, because of the potential for misidentification ofdibromochloromethane (DBCM) as EDB.504.1-23.0DEFINITIONS3.1Laboratory Duplicates (LD1 and LD2) -- Two aliquots of the same sample takenin the laboratory and analyzed separately with identical procedures. Analyses ofLD1 and LD2 indicate the precision associated with laboratory procedures, butnot with sample collection, preservation, or storage procedures.3.2Field Duplicates (FD1 and FD2) -- Two separate samples collected at the sametime and place under identical circumstances and treated exactly the samethroughout field and laboratory procedures. Analyses of FD1 and FD2 give ameasure of the precision associated with sample collection, preservation andstorage, as well as with laboratory procedures.3.3Laboratory Reagent Blank (LRB) -- An aliquot of reagent water or other blankmatrix that is treated exactly as a sample including exposure to all glassware,equipment, solvents, reagents, internal standards, and surrogates that are usedwith other samples. The LRB is used to determine if method analytes or otherinterferences are present in the laboratory environment, the reagents, or theapparatus.3.4Field Reagent Blank (FRB) -- An aliquot of reagent water or other blank matrixthat is placed in a sample container in the laboratory and treated as a sample inall respects, including shipment to the sampling site, exposure to sampling siteconditions, storage, preservation and all analytical procedures. The purpose ofthe FRB is to determine if method analytes or other interferences are present inthe field environment.3.5Instrument Performance Check Solution (IPC) -- A solution of one or moremethod analytes, surrogates, internal standards, or other test substances used toevaluate the performance of the instrument system with respect to a defined setof criteria.3.6Laboratory Fortified Blank (LFB) -- An aliquot of reagent water or other blankmatrix to which known quantities of the method analytes are added in thelaboratory. The LFB is analyzed exactly like a sample, and its purpose is todetermine whether the methodology is in control, and whether the laboratory iscapable of making accurate and precise measurements.3.7Laboratory Fortified Sample Matrix (LFM) -- An aliquot of an environmentalsample to which known quantities of the method analytes are added in thelaboratory. The LFM is analyzed exactly like a sample, and its purpose is todetermine whether the sample matrix contributes bias to the analytical results.The background concentrations of the analytes in the sample matrix must bedetermined in a separate aliquot and the measured values in the LFM correctedfor background concentrations.504.1-33.8Stock Standard Solution -- A concentrated solution containing one or methodanalytes prepared in the laboratory using assayed reference materials orpurchased from a reputable commercial source.3.9Primary Dilution Standard Solution (PDS) -- A solution of several analytesprepared in the laboratory from stock standard solutions and diluted as neededto prepare calibration solutions and other needed analyte solutions.3.10Calibration Standard (CAL) -- A solution prepared from the primary dilutionstandard solution and stock standard solutions of the internal standards andsurrogate analytes. The CAL solutions are used to calibrate the instrumentresponse with respect to analyte concentration.3.11Quality Control Sample (QCS) -- A solution of method analytes of knownconcentrations that is used to fortify an aliquot of LRB or sample matrix. TheQCS is obtained from a source external to the laboratory and different from thesource of calibration standards. It is used to check laboratory performance withexternally prepared test materials.3.12Procedural Standard Calibration -- A calibration method where aqueouscalibration standards are prepared and processed (e.g., purged,extracted, and/orderivatized) in exactly the same manner as a sample. All steps in the processfrom addition of sampling preservatives through instrumental analyses areincluded in the calibration. Using procedural standard calibration compensatesfor any inefficiencies in the processing procedure.4.0INTERFERENCES4.1Impurities contained in the extracting solvent usually account for the majority ofthe analytical problems. Solvent blanks should be analyzed on each new bottleof solvent before use. Indirect daily checks on the extracting solvent are obtainedby monitoring the reagent water blanks (Section 7.2.4). Whenever an interferenceis noted in the reagent water blank, the analyst should reanalyze the extractingsolvent. Low level interferences generally can be removed by distillation or3column chromatography. When a solvent is purified, preservatives put into thesolvent by the manufacturer are removed thus potentially making the shelf-lifeshort. It is generally more economical to obtain a new source of solvent.Interference-free solvent is defined as a solvent containing less than the MDL ofan individual analyte interference. Protect interference-free solvents by storingin an area free of organochlorine solvents.4.2This liquid/liquid extraction technique efficiently extracts a wide boiling rangeof non-polar organic compounds and, in addition, extracts polar organiccomponents of the sample with varying efficiencies.4.3Dibromochloromethane is a common disinfection byproduct in chlorinateddrinking waters that frequently occurs at relatively high concentrations. DBCMcan elute very close to EDB, and a high concentration of DBCM may mask a low504.1-4concentration of EDB, or be misidentified as EDB. Therefore, special care shouldbe taken in the identification and confirmation of EDB.4.4It is important that samples and working standards be contained in the samesolvent. The solvent for working standards must be the same as the final solventused in sample preparation. If this is not the case, chromatographic comparabilityof standards to sample may be affected.5.0SAFETY5.1The toxicity and carcinogenicity of chemicals used in this method have not beenprecisely defined; each chemical should be treated as a potential health hazard,and exposure to these chemicals should be minimized. Each laboratory isresponsible for maintaining awareness of OSHA regulations regarding safehandling of chemicals used in this method. Additional references to laboratory5-7safety are available for the information of the analyst.5.2EDB, DBCP, and 123TCP have all been tentatively classified as known orsuspected human or mammalian carcinogens. Pure standard materials and stockstandard solutions of these compounds should be handled in a hood or glovebox.A NIOSH/MESA approved toxic gas respirator should be worn when the analysthandles high concentrations of these toxic compounds.6.0EQUIPMENT AND SUPPLIES (All specifications are suggested. Catalog numbers areincluded for illustration only.)6.1Sample Containers -- 40 mL screw cap vials each equipped with a Teflon-linedcap. Individual vials shown to contain at least 40.0 mL can be calibrated at the35.0 mL mark so that volumetric, rather than gravimetric, measurements ofsample volumes can be performed. Prior to use, wash vials and septa withdetergent and rinse with tap and distilled water. Allow the vials and septa to airdry at room temperature, place in a 105°C oven for one hour, then remove andallow to cool in an area free of organic solvent vapors.6.2Vials -- auto sampler, screw cap with Teflon faced septa, 1.8 mL.6.3Micro Syringes -- 10 µL, 25 µL, and 100 µL.6.4Pipettes -- 2.0 mL and 5.0 mL transfer.6.5Standard Solution Storage Containers -- 15-mL bottles with Teflon lined screwcaps.6.6Gas Chromatography System504.1-56.6.1The gas chromatograph must be capable of temperature programming andshould be equipped with a linearized electron capture detector and acapillary column split/splitless injector.6.6.2Two gas chromatography columns are recommended. Column A providesseparation of the method analytes without interferences fromtrihalomethanes (Section 4.3). Column A should be used as the primaryanalytical column unless routinely occurring analytes are not adequatelyresolved. Column B is recommended for use as a confirmatory columnwhen GC/MS confirmation is not viable. Retention times for the methodanalytes on these columns are presented in Table 1.6.6.3Column A (Primary Column) -- DB-1, 30 m x 0.25 mm ID, 1.0 µm filmthickness fused silica capillary column or equivalent. The linear velocityof the helium carrier gas should be about 25 cm/sec at 100°C. Thecolumn temperature is programmed to hold at 40°C for four minutes, toincrease to 240°C at 10°C/min., and hold at 240°C for five minutes or untilall expected compounds have eluted.6.6.4Column B (Alternative Column) -- DB-624, 30 m x 0.32 mm ID, 1.8 µmfilm thickness fused silica capillary column or equivalent. The linearvelocity of the helium carrier gas should be about 25 cm/sec at 100°C.The column temperature is programmed as described in Section 6.6.3.7.0REAGENTS AND STANDARDS7.1Reagents7.1.1Hexane Extraction Solvent, UV Grade -- Distilled in glass7.1.2Methyl Alcohol, ACS Reagent Grade -- Demonstrated to be free of methodanalytes above the MDLs.7.1.3Sodium Chloride, NaCl, ACS Reagent Grade - For pretreatment beforeuse, pulverize a batch of NaCl and place in a muffle furnace at roomtemperature. Increase the temperature to 400°C for 30 minutes. Place ina bottle and cap.7.1.4Sodium Thiosulfate, Na S O,ACS Reagent Grade -- For preparation of223solution (40 mg/mL), dissolve 1 g of Na S O in reagent water and bring223to 25 mL volume in a volumetric flask.504.1-67.2Reagent Water -- Reagent water is defined as water free of interferences above theanalyte MDLs.7.2.1Reagent water can be generated by passing tap water through a filter bedcontaining activated carbon. Change the activated carbon when there isevidence that volatile organic compounds are breaking through thecarbon.7.2.2 A Millipore Super-Q Water System or its equivalent may be used togenerate deionized reagent water.7.2.3Reagent water may also be prepared by boiling water for 15 minutes.Subsequently, while maintaining the temperature at 90°C, bubble acontaminant-free inert gas through the water at 100 mL/min. forone hour. While still hot, transfer the water to a narrow mouth screw capbottle with a Teflon seal.7.2.4Test reagent water each day it is used by analyzing it according toSection 11.0.7.3Stock Standard Solutions (SSS) -- These solutions may be purchased as certifiedsolutions or prepared from pure standard materials using the following procedures:7.3.1Place about 9.8 mL of methanol into a 10 mL ground-glass stopperedvolumetric flask. Allow the flask to stand, unstoppered, for about10 minutes and weigh to the nearest 0.1 mg.7.3.2Use a 100 µL syringe and immediately add two or more drops of standardmaterial to the flask. Be sure that the standard material falls directly intothe methanol without contacting the neck of the flask.7.3.3Reweigh, dilute to volume, stopper, then mix by inverting the flask severaltimes. Calculate the concentration in micrograms per microliter from thenet gain in weight.7.3.4Store stock standard solutions in 15 mL bottles equipped with Teflon linedscrew caps. Methanol solutions prepared from liquid analytes are stablefor at least four weeks when stored at 4°C.7.4Primary Dilution Standard Solutions (PDS) -- Use stock standard solutions toprepare primary dilution standard solutions that contain all three analytes in methanol. The primary dilution standards should be prepared at concentrations that can be easily diluted to prepare aqueous calibration standards (Section 10.1.1) that will bracket the working concentration range. Store the primary dilution standard solutions with minimal headspace and check frequently for signs of deterioration or evaporation, especially just before preparing calibration504.1-7standards. The storage time described for stock standard solutions in Section7.3.4 also applies to primary dilution standard solutions.7.5Laboratory Fortified Blank (LFB), Sample Concentrate (0.25 µg/mL) -- Prepare anLFB sample concentrate of 0.25 µg/mL of each analyte from the stock standardsolutions prepared in Section 7.3.7.6MDL Check Sample Concentrate (0.02 µg/mL) -- Dilute 2 mL of LFB sampleconcentrate (Section 7.5) to 25 mL with methanol.8.0SAMPLE COLLECTION, PRESERVATION, AND STORAGE8.1Sample Collection8.1.1Replicate Field Reagent Blanks (FRB) -- Must be handled along with eachsample set, which is composed of the samples collected from the samegeneral sampling site at approximately the same time. At the laboratory,fill a minimum of two sample bottles with reagent water, seal, and shipto the sampling site along with sample bottles. Wherever a set of samplesis shipped and stored, it must be accompanied by the FRB.8.1.2Collect all samples in 40 mL bottles into which 3 mg of sodium thiosulfatecrystals have been added to the empty bottles just prior to shipping to thesampling site. Alternatively, 75 µL of freshly prepared sodium thiosulfatesolution (40 mg/mL) may be added to empty 40 mL bottles just prior tosample collection. This dechlorinating agent must be added to eachsample to avoid the possibility of reactions that may occur betweenresidual chlorine and indeterminant contaminants present in somesolvents, yielding compounds that may subsequently interfere with theanalysis. The presence of sodium thiosulfate will arrest further formationof DBCM (See Section 4.3).8.1.3When sampling from a water tap, open the tap and allow the system toflush until the water temperature has stabilized (usually about10 minutes). Adjust the flow to about 500 mL/min. and collect samplesfrom the flowing stream.8.1.4When sampling from a well, fill a wide-mouth bottle or beaker withsample, and carefully fill 40 mL sample bottles.8.2Sample Preservation8.2.1The samples must be chilled to 4°C or less at the time of collection andmaintained at that temperature until analysis. Field samples that will notbe received at the laboratory on the day of collection must be packagedfor shipment with sufficient ice to ensure that they will be ≤4°C on arrivalat the laboratory.8.3Sample Storage504.1-88.3.1Store samples and field reagent blanks together at 4°C until analysis. Thesample storage area must be free of organic solvent vapors.8.3.2Because 1,2,3-trichloropropane has been added to the analyte list in thismethod and has been found to have a 14-day maximum holding time in4studies conducted for Method 524.2, all samples must be extracted within14 days of collection. Samples not analyzed within this period must bediscarded and replaced. Because of the potential for solvent evaporation,it is preferred that extracts be analyzed immediately followingpreparation. When necessary, extracts may be stored in tightly cappedvials (Section 6.2) at 4°C or less for up to 24 hours.9.0QUALITY CONTROL9.1Each laboratory that uses this method is required to operate a formal qualitycontrol program. The minimum requirements of this program consist of an initialdemonstration of laboratory capability and an ongoing analysis of field reagentblanks (FRB), laboratory reagent blanks (LRB), laboratory fortified blanks (LFB),laboratory fortified sample matrix (LFM), and quality control samples (QCS) toevaluate and document data quality. Ongoing data quality checks are comparedwith established performance criteria to determine if the results of analyses meetthe performance characteristics of the method.9.1.1The analyst must make an initial determination of the method detectionlimits and demonstrate the ability to generate acceptable precision withthis method. This is established as described in Section 9.2.9.1.2In recognition of advances that are occurring in chromatography, theanalyst is permitted certain options to improve the separations or lowerthe cost of measurements. Each time such a modification is made to themethod, the analyst is required to repeat the procedure in Section 9.2.9.1.3Each day, the analyst must analyze a laboratory reagent blank (LRB) anda field reagent blank, if applicable (Section 8.1.1), to demonstrate thatinterferences from the analytical system are under control before anysamples are analyzed. In general, background interferences co-elutingwith method analytes should be below the method detection limits.9.1.4The laboratory must, on an ongoing basis, demonstrate through theanalyses of laboratory fortified blanks (LFB) that the operation of themeasurement system is in control. This procedure is described inSection 9.3. The frequency of the LFB analyses is equivalent to 10% of allsamples analyzed.9.1.5The laboratory should demonstrate the ability to analyze low levelsamples weekly. The procedure for low level LFB samples is described inSection 9.4.504.1-99.2Initial Demonstration of Capability9.2.1Prepare four to seven LFBs at a concentration equal to 10 times the MDLor at a concentration in the middle of the calibration range established inSection 10.0.9.2.2Analyze the LFBs according to the method beginning in Section 11.0.9.2.3Calculate the mean concentration found (X) in µg/L, and the standarddeviation of the concentrations in µg/L, for each analyte.9.2.4For each analyte, X should be between 70% and 130% of the true value.The RSD should be 20% or less. If the results for all three analytes meetthese criteria, the system performance is acceptable. If any analyte failsto meet the criteria, correct the source of the problem and repeat the test.Caution: No attempts to establish low detection limits should be madebefore instrument optimization and adequate conditioning of both thecolumn and the GC system. Conditioning includes the processing of LFBand LFM samples containing moderate analyte concentrations.9.2.5Determination of MDL -- Prepare four to seven LFBs at a lowconcentration. Use the concentrations in Tables 2 and 3 as a guideline, oruse calibration data obtained (Section 10.0) to estimate a concentration foreach analyte that will produce a chromatographic peak with a three to fivesignal to noise ratio. It is recommended that LFBs for determination ofthe MDL be prepared and analyzed over a period of several days, so thatday to day variations will be reflected in the precision data.9.2.6Analyze the LFBs as directed in Section 11.0. Calculate the mean amountrecovered and the standard deviation of these measurements. Use thestandard deviation and the equation in Section 13.0 to calculate the MDL.9.3Assessing Laboratory Performance -- The laboratory must demonstrate that themeasurement system is in control by analyzing an LFBs of the analytes at0.25 µg/L concentration level. This must be demonstrated on a frequencyequivalent to 10% of the sample load, or one per batch of samples extracted, whichever is greater.9.3.1Prepare an LFB sample (0.25 µg/L) by adding 35 µL of LFB concentrate(Section 7.5) to 35 mL of reagent water in a 40 mL bottle.9.3.2Immediately analyze the LFB sample according to Section 11.0 andcalculate the recovery for each analyte. The recovery should be between70% and 130% of the expected value.9.3.3If the recovery for either analyte falls outside the designated range, theanalyte fails the acceptance criteria. A second LFB containing each analyte504.1-10that failed must be analyzed. Repeated failure, however, will confirm ageneral problem with the measurement system. If this occurs, locate andcorrect the source of the problem and repeat the test.9.3.4Since this LFB is prepared in the same manner as a calibration verificationstandard, this LFB data can also be used to satisfy the calibrationrequirement in Section 10.1.4.9.4Assessing Laboratory Sensitivity -- The laboratory should demonstrate the abilityto analyze low level samples for EDB and DBCP weekly.9.4.1Prepare an MDL check sample (a LFB fortified at 0.02 µg/L) andimmediately analyze according to the method in Section 11.0.9.4.2The instrument response must indicate that the laboratory's MDL isdistinguishable from instrument background signal. If it is not, correct theproblem (increase sensitivity) and repeat Section 9.4.1.9.4.3For each analyte, the recovery must be between 60% and 140% of theexpected value. These criteria are looser than those in Sections 9.2.4 and9.3.2 because of the low concentration.9.4.4When either analyte fails the test, the analyst should repeat the test forthat analyte. Repeated failure, however, will confirm a general problemwith the measurement system or faulty samples and/or standards. If thisoccurs, locate and correct the source of the problem and repeat the test.9.5Assessing Matrix Effects -- At least once in every 20 samples, fortify an aliquotof a randomly selected routine sample with known amounts of the analytes. The added concentration should not be less than the background concentration of the sample selected for fortification. To simplify these checks, it would be convenient to use LFM concentrations ≈10X MDL. Over time, recovery should be evaluated on fortified samples from all routine sources. Calculate the percent recovery (R)i for each analyte, corrected for background concentrations measured in the unfortified sample. If the recovery of any such analyte falls outside the range of ±35% of the fortified amount, and the laboratory performance for that analyte is shown to be in control (Section 9.3), the recovery problem encountered with the dosed sample is judged to be matrix related, not system related. The result for that analyte in the unfortified sample is labeled suspect/matrix to inform the data user that the results are suspect due to matrix effects.9.6It is highly recommended that a laboratory establish its ability todistinguish DBCM from EDB. This is particularly important if samplesfrom chlorinated sources or unfamiliar sources are to be analyzed(Section 4.3). Standards of DBCM should be analyzed periodically toestablish its retention time relative to that of EDB. When evaluating thisretention time difference, the analyst should keep in mind that DBCM islikely to be present in concentrations much larger than EDB, and that the504.1-11ability to detect EDB may deteriorate with increasing DBCMconcentration.9.7At least quarterly, a quality control sample (QCS) should be analyzed. Ifmeasured analyte concentrations are not of acceptable accuracy, check the entireanalytical procedure to locate and correct the problem source.9.8It is recommended that the laboratory adopt additional quality assurance practicesfor use with this method. The specific practices that are most productive dependupon the needs of the laboratory and the nature of the samples. Field duplicatesmay be analyzed to assess the precision of the environmental measurements.Whenever possible, the laboratory should analyze standard reference materialsand participate in relevant performance evaluation studies.10.0CALIBRATION AND STANDARDIZATION10.1Calibration and Standardization10.1.1At least three calibration standards are needed; five are recommended.Guidance on the number of standards is as follows: A minimum of threecalibration standards are required to calibrate a range of a factor of 20 inconcentration. For a factor of 50 use at least four standards, and for afactor of 100 at least five standards. The lowest standard should representanalyte concentrations near, but above, their respective MDLs. Theremaining standards should bracket the analyte concentrations expectedin the sample extracts, or should define the working range of the detector.10.1.2To prepare a calibration standard (CAL), add an appropriate volume ofa primary dilution standard solution to an aliquot of reagent water in avolumetric flask. If <20 µL of a standard is added to the reagent water,poor precision may result. Use a 25 µL micro syringe and rapidly injectthe alcoholic standard into the expanded area of the filled volumetricflask. Remove the needle as quickly as possible after injection. Mix byinverting the flask several times. Discard the contents contained in theneck of the flask. Aqueous standards should be prepared fresh andextracted immediately after preparation unless sealed and stored withoutheadspace as described in Section 8.0. Alternatively, measure a 35 mLvolume of reagent water in a 50 mL graduated cylinder and transfer it toa 40 mL sample container (Section 6.1). Use a micro syringe to inject thestandard into the reagent water. Cap and mix gently.10.1.3Analyze each calibration standard according to Section 11.0 and record thepeak height or area response from each standard. Create a calibrationcurve by plotting peak area response versus the concentration in thestandard. Alternatively, if the ratio of concentration to response(calibration factor) is a constant over the working range (<20% relativestandard deviation), linearity through the origin can be assumed and the504.1-12。