中药新药研发流程图
- 格式:doc
- 大小:22.31 KB
- 文档页数:3


中药复方新药研发标准操作规程(一药学)----------------房树标初拟于2009年7月25日星期六--------------中药复方新药开发流程草图:立项选题:3.立题目的与依据,病人发病率、市场、疾病发病机制,目前治疗进展,可能开发的治疗药物临床疗效及其作用机制,病理模型与检测指标、临床病例采集难易,药理临床费用,原料辅料供应情况,制剂工艺,新药成本,研发周期及风险控制。
前期要做配方药效筛选,同步考察毒副作用等以减少开发风险。
8.药材来源及鉴定依据1、药材或饮片,提取物(少用):供货方资质证明、购销合同、发票、产品质检报告或与质量标准。
2、物流入库登记,质检科检验产品并出具检验报告。
3、检验项目参照新版药典各药材项,包括:性状(外观、气味等)、鉴别(经验、理化、显微)、检查(水分、灰分、杂质、毒性成分、重金属、有害元素、农残)、含量测定或浸出物测定。
9、10、11三项原料药材符合法定标准的可不提供。
12.生产工艺研究资料,工艺验证资料及文献资料,辅料来源及质量标准。
1.(12与13文献资料):文献检索包括复方中各位药材的成分、理化性质、含量测定方法、药理、药代、临床,新药制剂工艺等全面信息,为以后的药学工艺、质量标准的制定,药理模型的建立、临床方案的设计奠定科研基础。
2.辅料来源及质量标准:供货方资质证明、购销合同、发票、产品质检报告与质量标准。
物流入库登记,质检检验产品并出具检验报告。
检验项目参照新版药典各辅料项。
3.生产工艺研究资料,工艺验证资料:购进原料后,现通过实验室摸索(正交设计等摸索提取及制剂工艺)再通过实验室小试建立工艺。
注意:工艺指标选择要有意义(有效成分或指标成分),小试工艺参数最好是个范围,以便于生产条件的实现,同时也要有一个最佳工艺参数。
如提取工艺参数:溶剂量,时间、温度;直接浓缩至一定密度的浸膏,考察出膏量及有效成分含量。
可进一步考察浓缩参数:温度、压力、时间;指标可选膏的浓度。

中药新药开发步骤和周期新药申请从研发到生产上市,需要漫长时间才能获批,而这几年医药市场瞬息万变,多种不确定原因很之大,所以能够说,新药研发是一项开发周期长、资金投入大、不可估计原因多系统工程,含有较高风险性。
通常药品研发大致步骤及对应花费时间以下:立项(4-6月)→临床前研究(18-36月)→CDE待批临床(大约1年)→临床试验(3年左右)→CDE待批生产(1年-n年)→批文生产转移(约6个月)总计:最少8年以上注:CDE国家食品药品监督管理局药品审评中心英文缩写,其职能关键对新药、仿制药、补充申请等进行审评。
具体以下:1、立项:简单说在确定开发某品种前,需要进行一系列市场调研工作,经过对市场,流行病学,技术,疗效和安全,知识产权,成品成本,国家政策,企业本身条件等方面考察,来确定研发品种或诊疗某类疾病药品上市后市场潜力,从而选择适合本企业品种。
通常需要多个月时间来立项。
立项决定了该品种未来市场,所以立项至关关键。
2、临床前研究:通常对于中药品种研发步骤以下:小试产品→初步药效筛选→化学成份研究→处方、药效筛选→药理毒理→药剂工艺制备研究→质量标准→中试放大→稳定性试验→资料整理报批因为这个过程是从发觉药用原料潜在价值到确定其开发价值过程(成药性研究),需要对要用原料做大量多学科基础研究,这个过程需要1-2年;进入工艺制剂、质量标准和稳定性试验,即使最简单品种通常也需要12-24月研究过程;所以基础全部需要24个月到36个月时间研究。
该阶段可能影响时间关键在于原料化学成份研究和药效学研究,不确定原因较多,是否顺利决定了时间长短。
药品临床前研发,一定要研究透彻,不仅利于申报后尽可能少发补资料甚至不发补,立即获批,更有利于未来获批后顺利生产上市。
3、CDE待批临床:依据药品注册管理措施相关要求,省局30日内完成资料形式审查,注册现场核查等,但可能因为补充资料等等事宜,往往时间会超出30天。
国家局CDE:新药临床试验:90日;获准进入特殊审批程序品种:80日。


中药新药研发申报流程及相关申报材料说明一、中药新药的注册分类及说明1.1注册分类中药新药注册按审批管理的要求分以下几类:1.未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。
2.新发现的药材及其制剂.3.新的中药材代用品。
4.药材新的药用部位及其制剂。
5.未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。
6.未在国内上市销售的中药、天然药物复方制剂。
7.改变国内已上市销售中药、天然药物给药途径的制剂。
8.改变国内已上市销售中药、天然药物剂型的制剂.9.仿制药。
1.2说明注册分类1-6的品种为新药,注册分类7、8按新药申请程序申报,注册分类9的品种为已有国家标准的药品。
二、中药新药的研发及申报流程中药新药的研发申报一般按以下程序进行:选题立项——临床前研究-—临床研究—-申报审批-—正式生产,其中,新药临床前及临床研究的主要内容及注意事项分别列举如下:2.1 新药的临床前研究(一)主要内容:新药的临床前研究主要包括制备工艺(中药制剂包括原药材的来源、加工及炮制)、理化性质、纯度、检验方法、处方筛选、剂型、稳定性、质量标准、药理、毒理、动物药代动力学等研究。
新发现中药材还应包括来源、生态环境、栽培(养殖)技术、采收处理、加工炮制等研究。
(二)注意事项:从事新药安全性研究的实验室应符合国家药品监督管理局《药品非临床研究质量管理规范》(GLP)的相应要求,实验动物应符合国家药品监督管理局的有关要求,以保证各项实验的科学性和实验结果的可靠性.2。
2 新药的临床研究(一)主要内容:新药的临床研究包括临床试验和生物等效性试验.新药的临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。
Ⅰ期临床试验:初步的临床药理学及人体安全性评价试验。
观察人体对于新药的耐受程度和药物代谢动力学,为制定给药方案提供依据。
Ⅱ期临床试验:随机盲法对照临床试验。
对新药有效性及安全性作出初步评价,推荐临床给药剂量.Ⅲ期临床试验:扩大的多中心临床试验。



中药新药研发申报流程及相关申报材料说明一、中药新药的注册分类及说明1.1注册分类中药新药注册按审批管理的要求分以下几类:1.未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。
2.新发现的药材及其制剂。
3.新的中药材代用品.4.药材新的药用部位及其制剂。
5.未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。
6.未在国内上市销售的中药、天然药物复方制剂。
7.改变国内已上市销售中药、天然药物给药途径的制剂。
8.改变国内已上市销售中药、天然药物剂型的制剂。
9.仿制药.1.2说明注册分类1-6的品种为新药,注册分类7、8按新药申请程序申报,注册分类9的品种为已有国家标准的药品.二、中药新药的研发及申报流程中药新药的研发申报一般按以下程序进行:选题立项——临床前研究——临床研究——申报审批——正式生产,其中,新药临床前及临床研究的主要内容及注意事项分别列举如下:2.1 新药的临床前研究(一)主要内容:新药的临床前研究主要包括制备工艺(中药制剂包括原药材的来源、加工及炮制)、理化性质、纯度、检验方法、处方筛选、剂型、稳定性、质量标准、药理、毒理、动物药代动力学等研究。
新发现中药材还应包括来源、生态环境、栽培(养殖)技术、采收处理、加工炮制等研究。
(二)注意事项:从事新药安全性研究的实验室应符合国家药品监督管理局《药品非临床研究质量管理规范》(GLP)的相应要求,实验动物应符合国家药品监督管理局的有关要求,以保证各项实验的科学性和实验结果的可靠性。
2.2 新药的临床研究(一)主要内容:新药的临床研究包括临床试验和生物等效性试验。
新药的临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。
Ⅰ期临床试验:初步的临床药理学及人体安全性评价试验。
观察人体对于新药的耐受程度和药物代谢动力学,为制定给药方案提供依据。
Ⅱ期临床试验:随机盲法对照临床试验。
对新药有效性及安全性作出初步评价,推荐临床给药剂量。
Ⅲ期临床试验:扩大的多中心临床试验.应遵循随机对照原则,进一步评价有效性、安全性。

中药新药开发流程和周期新药申请从研发到生产上市,需要漫长的时间才能获批,而这几年的医药市场瞬息万变,各种不确定因素非常之大,因此可以说,新药研发是一项开发周期长、资金投入大、不可预测因素多的系统工程,具有较高的风险性。
一般药品研发的大致流程及相应花费时间如下:立项(4-6月)→临床前研究(18-36月)→CDE待批临床(大约1年)→临床试验(3年左右)→CDE待批生产(1年-n年)→批文生产转移(约6个月)总计:至少8年以上注:CDE国家食品药品监督管理局药品审评中心英文缩写,其职能主要对新药、仿制药、补充申请等进行审评。
具体如下:1、立项:简单的说在确定开发某品种前,需要进行一系列市场调研工作,通过对市场,流行病学,技术,疗效和安全,知识产权,成品成本,国家政策,企业自身条件等方面的考察,来确定研发品种或治疗某类疾病药物上市后的市场潜力,从而选择适合本企业的品种。
一般需要几个月时间来立项。
立项决定了该品种的将来市场,因此立项至关重要。
2、临床前研究:一般对于中药品种的研发流程如下:小试产品→初步药效筛选→化学成分研究→处方、药效筛选→药理毒理→药剂工艺制备研究→质量标准→中试放大→稳定性实验→资料整理报批由于这个过程是从发现药用原料的潜在价值到确认其开发价值的过程(成药性研究),需要对要用原料做大量的多学科基础研究,这个过程需要1-2年;进入工艺制剂、质量标准和稳定性试验,即使最简单的品种一般也需要12-24月研究过程;因此基本都需要24个月到36个月时间研究。
该阶段可能影响时间的关键在于原料的化学成分研究和药效学研究,不确定因素较多,是否顺利决定了时间的长短。
药物临床前研发,一定要研究透彻,不仅利于申报后尽可能少的发补资料甚至不发补,尽快获批,更有利于将来获批后顺利生产上市。
3、CDE待批临床:根据药品注册管理办法相关规定,省局30日内完成资料的形式审查,注册现场核查等,但可能由于补充资料等等事宜,往往时间会超过30天。

中药新药研发申报流程及相关申报材料说明一、中药新药的注册分类及说明1.1注册分类中药新药注册按审批管理的要求分以下几类:1.未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。
2.新发现的药材及其制剂。
3.新的中药材代用品。
4.药材新的药用部位及其制剂。
5.未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。
6.未在国内上市销售的中药、天然药物复方制剂。
7.改变国内已上市销售中药、天然药物给药途径的制剂。
8.改变国内已上市销售中药、天然药物剂型的制剂。
9.仿制药。
1.2说明注册分类1-6的品种为新药,注册分类7、8按新药申请程序申报,注册分类9的品种为已有国家标准的药品。
二、中药新药的研发及申报流程中药新药的研发申报一般按以下程序进行:选题立项——临床前研究——临床研究——申报审批——正式生产,其中,新药临床前及临床研究的主要内容及注意事项分别列举如下:2.1 新药的临床前研究(一)主要内容:新药的临床前研究主要包括制备工艺(中药制剂包括原药材的来源、加工及炮制)、理化性质、纯度、检验方法、处方筛选、剂型、稳定性、质量标准、药理、毒理、动物药代动力学等研究。
新发现中药材还应包括来源、生态环境、栽培(养殖)技术、采收处理、加工炮制等研究。
(二)注意事项:从事新药安全性研究的实验室应符合国家药品监督管理局《药品非临床研究质量管理规范》(GLP)的相应要求,实验动物应符合国家药品监督管理局的有关要求,以保证各项实验的科学性和实验结果的可靠性。
2.2 新药的临床研究(一)主要内容:新药的临床研究包括临床试验和生物等效性试验。
新药的临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。
Ⅰ期临床试验:初步的临床药理学及人体安全性评价试验。
观察人体对于新药的耐受程度和药物代谢动力学,为制定给药方案提供依据。
Ⅱ期临床试验:随机盲法对照临床试验。
对新药有效性及安全性作出初步评价,推荐临床给药剂量。
Ⅲ期临床试验:扩大的多中心临床试验。
中药复方新药研发标准操作规程(一药学)
----------------房树标初拟于2009年7月25日星期六--------------
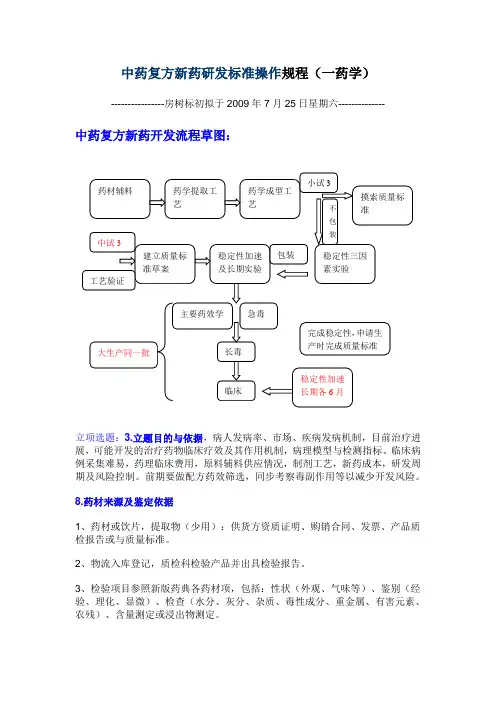
中药复方新药开发流程草图:
立项选题:3.立题目的与依据,病人发病率、市场、疾病发病机制,目前治疗进展,可能开发的治疗药物临床疗效及其作用机制,病理模型与检测指标、临床病例采集难易,药理临床费用,原料辅料供应情况,制剂工艺,新药成本,研发周期及风险控制。
前期要做配方药效筛选,同步考察毒副作用等以减少开发风险。
8.药材来源及鉴定依据
1、药材或饮片,提取物(少用):供货方资质证明、购销合同、发票、产品质检报告或与质量标准。
2、物流入库登记,质检科检验产品并出具检验报告。
3、检验项目参照新版药典各药材项,包括:性状(外观、气味等)、鉴别(经验、理化、显微)、检查(水分、灰分、杂质、毒性成分、重金属、有害元素、农残)、含量测定或浸出物测定。
9、10、11三项原料药材符合法定标准的可不提供。
12.生产工艺研究资料,工艺验证资料及文献资料,辅料来源及质量标准。
1.(12与13文献资料):文献检索包括复方中各位药材的成分、理化性质、含量测定方法、药理、药代、临床,新药制剂工艺等全面信息,为以后的药学工艺、质量标准的制定,药理模型的建立、临床方案的设计奠定科研基础。
2.辅料来源及质量标准:供货方资质证明、购销合同、发票、产品质检报告与质量标准。
物流入库登记,质检检验产品并出具检验报告。
检验项目参照新版药典各辅料项。
3.生产工艺研究资料,工艺验证资料:购进原料后,现通过实验室摸索(正交设计等摸索提取及制剂工艺)再通过实验室小试建立工艺。
注意:工艺指标选择要有意义(有效成分或指标成分),小试工艺参数最好是个范围,以便于生产条件的实现,同时也要有一个最佳工艺参数。
如提取工艺参数:溶剂量,时间、温度;直接浓缩至一定密度的浸膏,考察出膏量及有效成分含量。
可进一步考察浓缩参数:温度、压力、时间;指标可选膏的浓度。
成型工艺参数:辅料种类、辅料量、制粒粒度、干燥温度,指标可选制粒成本与有效成分含量等。
生产车间中试及大生产验证工艺(车间要有批记录,质检要有检验报告)。
最后要做一个:工艺流程图。
14.质量研究工作的实验及文献资料,15.药品标准草案及起草说明,药品标准物质及有关资料(中国药品生物制品鉴定所)。
1.在前期小试及工艺摸索建立过程中所形成的初步质量标准,需要中试或以上产品(3批)的验证,质检科出具质检报告16.样品检验报告书,同时建立质量标准草案。
质量标准的内容及项目要求见附录(参照药典及新药研究技术要求及中药新药药学研究指南)
2.小试3批一是用来摸索质量标准,二是用来考察稳定性的影响因素实验(高温、高湿、光照各10天)(不要包装)。
同时做质量标准的3批中试及以上产品也可用来做稳定性加速6个月及长期实验(要求带包装样品)17.样品稳定性研究实验资料及文献资料。
18.直接接触药品的包装材料和容器的选择依据及质量标准。
影响因素做完后,根据实验结果选择包材(厂家资质证明,合同、发票、质量标准、质检报告),并用包装的样品做加速及长期稳定性实验,验证包装的安全、封藏性能;当然包材也要考虑便宜及美观性能。
7.药学研究资料综述
注:序号为注册申报材料序号。