usp31nf26s2_saccharin sodium
- 格式:pdf
- 大小:56.08 KB
- 文档页数:5

Fenofibrate非诺贝特lsopropyl2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoate---[49526-28-9]异硫氰酸酯 2 - [4 - (4 -氯)苯氧基] - 2 - 甲基丙醇--- [49526-28-9] Fenofibrate contains not less than 98.5 percent and more than 101.0 percent of C20H21CIO calculated on the dried basis非诺贝特含有不低于百分之98.5和超过百分之101.0 C20H21CIO Packaging and storage---包装和储存---Preserve in well-closed,light-resistant containers.Store at room temperature保存好封闭,光的容器。
在室温存放温度USP Reference standards---美国药典参考标准---USP Fenofibrate RS美国药典非诺贝特实寄存器USP Fenofibrate Related Compound A RS美国药典复方阿非诺贝特相关遥感USP Fenofibrate Related Compound B RS美国药典非诺贝特相关化合物B遥感USP Fenofibrate Related Compound C RS美国药典非诺贝特相关化合物C遥感Color of solution----溶液颜色----Reference solution----参考的解决方案----Mix 5 mL of Matching Fluid G and 95 mL of dilute hydrochloric acid (1in 40)混合5毫升在匹配液体G和在95毫升稀盐酸之中(1 比40)Test solution---测试解决方案---Transfer 500 mg of Fenofibrate to a 10-ml volumetric flask,dissolve in and dilute with acetone to volume,and mix转移至500毫克的非诺贝特10毫升容量瓶中,溶解并稀释丙酮体积和混合Procedure---程序---Proceed as directed under Color and Achromicity ;继续根据颜色和色度缺乏的指示;the Test solution is not more intensely colored than the Reference solution 测试方法是不是更激烈的参考解决方案比色Identification,Infrared Absorption识别,红外吸收Melting range,Class 1a ;between 79 and 82熔化范围,类别1a;79和82之间Acidity---Dissolve 1.0 g in 50 ml of alcohol previously neutralized to phenolphthalein TS,and titrate with 0.1 N sodium hydroxide VS;not more than 0.2 ml of 0.1 N sodium hydroxide vs is required to change the color of the indicator to pink1.0酸度---溶解在50毫升克酒精,以酚酞给付瓦解以前,和滴定0.1 N氢氧化钠VS;不超过0.2比0.1 N氢氧化钠毫升须改变为粉红色指示灯的颜色Loss on drying---干燥失重---Dry it in vacuum over phosphorus pentoxide at 60 to constant weight;it loses not more than 0.5% of its weight它比在真空干燥五氧化二磷在60至恒重,它失去了不超过其体重的0.5%Residue on ignition;not more than 0.1%,determined on 1.0 g灼烧残渣;不超过0.1%,对确定为1.0 gChloride 氯化物Test solution---Add 25 ml of water to 5.0g of Fenofibrate,and heat at 50 for 10 minutes测试解决方案---加25毫升水的非诺贝特5.0克,并在50加热10分钟Cool,dilute with water to 50.0 ml,filter,and use the filtrate凉的淡化水50.0毫升,过滤,用滤液[NOTE--Retain the remaining portion of the The solution for the test for Sulfate][注- 保留为硫酸盐]测试解决方案的其余部分Procedure---程序---Use 10 ml of the Test solution;使用10毫升的测试解决方案it shows no more chloride than corresponds to 0.15 ml of 0.020 N hydrochloric acid(0.01%)它表明没有更多的氯化物相当于0.15比0.020 N盐酸(0.01%毫升)Sulfate----硫酸盐---Use 10 ml of the Test solution retained from the test for Chloride;使用10毫升的测试解决方案的保留氯化物试验;it shows no more sulfate than corresponds to 0.15 ml of 0.020 N sulfuricacid (0.01%)它表明没有更多的硫酸相当于0.15 比0.020 N硫酸(0.01%毫升)Heavy metals,Method ;0.002%重金属,方法二; 0.002%Related compounds---相关化合物---Mobile phase---Proceed as directed in the Assay流动相---就如在检测指示Impurity standard solution---杂质标准溶液---Dissolve accurately weighed quantities of USP Fenofibrate RS,USP Fenofibrate Related Compound A RS,USP Fenofibrate Related Compound B RS,and USP Fenofibrate Related Compound C RS in Mobile phase,and dilute quantitatively,and stepwise if necessary精确称量溶解在流动相遥感非诺贝特美国药典,美国药典复方阿非诺贝特相关遥感,美国药典非诺贝特相关复合乙遥感,非诺贝特和USP遥感相关化合物C数量,稀释定量,并逐步的是必要的With Mobile phase to obtain a solution having known concentrations of about 1 per ml each of fenofibrate,fenofibrate related compound A,and fenofibrate related compound B,and about 2 per ml of fenofibrate related compound C随着流动相,以获得一个解决方案具有已知的约有百毫升的每个非诺贝特,非诺贝特相关化合物甲,乙和非诺贝特相关化合物的浓度,约占非诺贝特和相关化合物毫升2架CTest solution---测试解决方案---Prepare as directed for the Assay preparation为准备作为检测的筹备Chromatographic system(see Chromatography 621)---Proceed as directed in the Assay.色谱系统(见色谱621)---如在检测进行指导。
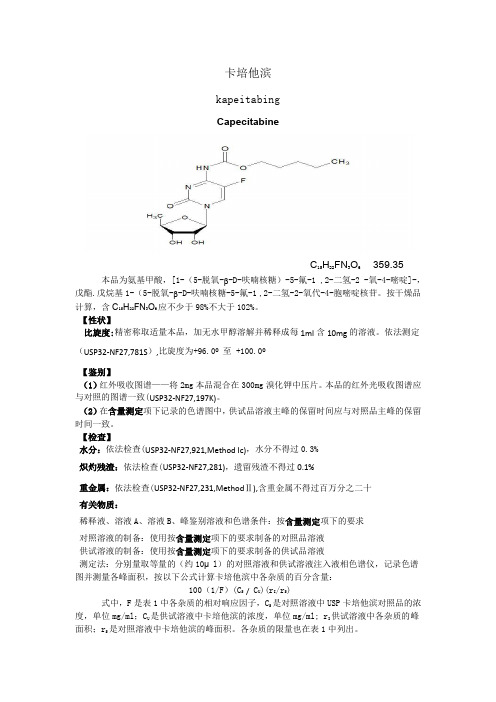
卡培他滨kapeitabingCapecitabineC15H22FN3O6 359.35本品为氨基甲酸,[1-(5-脱氧-β-D-呋喃核糖)-5-氟-1 ,2-二氢-2 -氧-4-嘧啶]-,戊酯.戊烷基1-(5-脱氧-β-D-呋喃核糖-5-氟-1 ,2-二氢-2-氧代-4-胞嘧啶核苷。
按干燥品计算,含C15H22FN3O6应不少于98%不大于102%。
【性状】比旋度:精密称取适量本品,加无水甲醇溶解并稀释成每1ml含10mg的溶液。
依法测定(USP32-NF27,781S),比旋度为+96.0⁰至 +100.0⁰【鉴别】(1)红外吸收图谱——将2mg本品混合在300mg溴化钾中压片。
本品的红外光吸收图谱应与对照的图谱一致(USP32-NF27,197K)。
(2)在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与对照品主峰的保留时间一致。
【检查】水分:依法检查(USP32-NF27,921,Method Ic),水分不得过0.3%炽灼残渣:依法检查(USP32-NF27,281),遗留残渣不得过0.1%重金属:依法检查(USP32-NF27,231,MethodⅡ),含重金属不得过百万分之二十有关物质:稀释液、溶液A、溶液B、峰鉴别溶液和色谱条件:按含量测定项下的要求对照溶液的制备:使用按含量测定项下的要求制备的对照品溶液供试溶液的制备:使用按含量测定项下的要求制备的供试品溶液测定法:分别量取等量的(约10μl)的对照溶液和供试溶液注入液相色谱仪,记录色谱图并测量各峰面积,按以下公式计算卡培他滨中各杂质的百分含量:100(1/F)(C S / C U)(r I/r S)式中,F是表1中各杂质的相对响应因子,C S是对照溶液中USP卡培他滨对照品的浓度,单位mg/ml;C U是供试溶液中卡培他滨的浓度,单位mg/ml; r I供试溶液中各杂质的峰面积;r S是对照溶液中卡培他滨的峰面积。
各杂质的限量也在表1中列出。

251 LEADThe imposition of stringent limits on the amounts of lead that may be present in pharmaceutical products has resulted in the use of two methods, of which the one set forth following depends upon extraction of lead by solutions of dithizone. For determination of the content of heavy metals generally, expressed as a lead equivalent, see Heavy Metals 231.Select all reagents for this test to have as low a content of lead as practicable, and store all reagent solutions in containers of borosilicate glass. Rinse thoroughly all glassware with warm dilute nitric acid (1 in 2), followed by water.Special Reagents—AMMONIA-CYANIDE SOLUTION— Dissolve 2 g of potassium cyanide in 15 mL of ammonium hydroxide, and dilute with water to 100 mL.AMMONIUM CITRATE SOLUTION— Dissolve 40 g of citric acid in 90 mL of water. Add 2 or 3 drops of phenol red TS, then cautiously add ammonium hydroxide until the solution acquires a reddish color. Remove any lead that may be present by extracting the solution with 20-mL portions of Dithizone Extraction Solution (see below), until the dithizone solution retains its orange-green color.DILUTED STANDARD LEAD SOLUTION— Dilute an accurately measured volume of StandardLead Solution (see Heavy Metals 231) [containing 10 µg of lead per mL], with 9 volumes of dilute nitric acid (1 in 100) to obtain a solution that contains 1 µg of lead per mL.DITHIZONE EXTRACTION SOLUTION— Dissolve 30 mg of dithizone in 1000 mL of chloroform, and add 5 mL of alcohol. Store the solution in a refrigerator.Before use, shake a suitable volume of the dithizone extraction solution with about half its volume of dilute nitric acid (1 in 100), discarding the nitric acid.HYDROXYLAMINE HYDROCHLORIDE SOLUTION— Dissolve 20 g of hydroxylamine hydrochloride in sufficient water to make approximately 65 mL. Transfer to a separator, add 5 drops of thymol blue TS, then add ammonium hydroxide until the solution assumes a yellow color. Add 10 mL of sodium diethyldithiocarbamate solution (1 in 25), mix, and allow to stand for 5 minutes. Extract this solution with successive 10- to 15-mL portions of chloroform until a 5-mL portion of the chloroform extract does not assume a yellow color when shaken with cupric sulfate TS. Add 3 N hydrochloric acid until the solution is pink (if necessary, add 1 or 2 drops more of thymol blue TS), and then dilute with water to 100 mL.POTASSIUM CYANIDE SOLUTION— Dissolve 50 g of potassium cyanide in sufficient water to make 100 mL. Remove the lead from this solution by extraction with successiveportions of Dithizone Extraction Solution, as described under Ammonium Citrate Solution above, then extract any dithizone remaining in the cyanide solution by shaking with chloroform. Finally dilute the cyanide solution with sufficient water so that each 100 mL contains 10 g of potassium cyanide.STANDARD DITHIZONE SOLUTION— Dissolve 10 mg of dithizone in 1000 mL of chloroform. Keep the solution in a glass-stoppered, lead-free bottle, suitably wrapped to protect it from light, and store in a refrigerator.Test Preparation— [NOTE—If, in the following preparation, the substance under test reacts too rapidly and begins charring with 5 mL of sulfuric acid before heating, use instead 10 mL of cooled dilute sulfuric acid (1 in 2), and add a few drops of the hydrogen peroxide before heating.] Where the monograph does not specify preparation of a solution, prepare a Test Preparation as follows. [Caution—Exercise safety precautions in this procedure, as some substances may react with explosive violence when digested with hydrogen peroxide. ] Transfer 1.0 g of the substance under test to a suitable flask, add 5 mL of sulfuric acid and a few glass beads, and digest on a hot plate in a hood until charring begins. Other suitable means of heating may be substituted. (Add additional sulfuric acid, if necessary, to wet the substance completely, but do not add more than a total of 10 mL.) Add, dropwise and with caution, 30 percent hydrogen peroxide, allowing the reaction to subside and again heating between drops. Add the first few drops very slowly, mix carefully to prevent a rapid reaction, and discontinue heating if foaming becomes excessive. Swirl the solution in the flask to prevent unreacted substance from caking on the walls of the flask. [NOTE—Add peroxide whenever the mixture turns brown or darkens.] Continue the digestion until the substance is completely destroyed, copious fumes of sulfur trioxide are evolved, and the solution is colorless. Cool, cautiously add 10 mL of water, evaporate until sulfur trioxide again is evolved, and cool. Repeat this procedure with another 10 mL of water to remove any traces of hydrogen peroxide. Cautiously dilute with 10 mL of water, and cool.Procedure— Transfer the Test Preparation, rinsing with 10 mL of water, or the volume of the prepared sample specified in the monograph to a separator, and, unless otherwise directed in the monograph, add 6 mL of Ammonium Citrate Solution and 2 mL of Hydroxylamine Hydrochloride Solution. (For the determination of lead in iron salts use 10 mL of Ammonium Citrate Solution.) Add 2 drops of phenol red TS, and make the solution just alkaline (red in color) by the addition of ammonium hydroxide. Cool the solution if necessary, and add 2 mL of Potassium Cyanide Solution. Immediately extract the solution with 5-mL portions of Dithizone Extraction Solution, draining off each extract into another separator, until the dithizone solution retains its green color. Shake the combined dithizone solutions for 30 seconds with 20 mL of dilute nitric acid (1 in 100), and discardthe chloroform layer. Add to the acid solution 5.0 mL of Standard Dithizone Solution and 4 mL of Ammonia-Cyanide Solution , and shake for 30 seconds: the color of the chloroform layer is of no deeper shade of violet than that of a control made with a volume of Diluted Standard Lead Solution equivalent to the amount of lead permitted in the sample under examination, and the same quantities of the same reagents and in the same manner as in the test with the sample.Auxiliary Information— Before contacting USP, have you checked for your question in the FAQs ?USP31–NF26 Page 135 Topic/Question Contact Expert Committee Monograph Kahkashan Zaidi, Ph.D.Senior Scientist1-301-816-8269(GC05) General Chapters 05。
![苯并呋喃酮衍生物用于治疗和预防糖尿病的用途[发明专利]](https://img.taocdn.com/s1/m/598e57c47375a417876f8fa7.png)
专利名称:苯并呋喃酮衍生物用于治疗和预防糖尿病的用途专利类型:发明专利
发明人:王英,安托恩·德·赛茨厄,高德·斯查勒,丹尼尔·德欧拉兹,丹尼尔·若德斯托弗,斯文·沃尔夫拉姆,彼得·韦伯,
桑德拉·泰克赛拉
申请号:CN200480013108.9
申请日:20040505
公开号:CN1787815A
公开日:
20060614
专利内容由知识产权出版社提供
摘要:本发明涉及一种化合物用作在哺乳动物中预防或治疗糖尿病的有效试剂的用途。
所述化合物选自苯并呋喃衍生物的组,其显示出很好的降血糖效果,因此是在哺乳动物中预防或治疗糖尿病的有效试剂。
申请人:帝斯曼知识产权资产管理有限公司
地址:荷兰海尔伦
国籍:NL
代理机构:北京东方亿思知识产权代理有限责任公司
代理人:肖善强
更多信息请下载全文后查看。

卡培他滨(USP31)C15H22FN3O6[].氨基甲酸,[1-(5-脱氧--D-呋喃核糖)-5-氟代-1,2-二氢-2-氧-4-嘧啶基]-, 戊基酯. 戊基 1-(5-脱氧--D-呋喃核糖)-5-氟代-1,2-二氢-2-氧-4-嘧啶氨基甲酸酯以无水和无溶剂物计,卡培他滨含C15H22FN3O6为%~%。
【包装和贮存】密闭,受控室温保存。
【性状】白色至类白色结晶性粉末。
易溶于甲醇,溶解于乙腈和乙醇,难溶于水。
【鉴别】A: 红外鉴别试样:2mg供试品与300mg溴化钾混合。
B:含量项下,样品溶液色谱图中主峰保留时间应与标准溶液色谱图中主峰保留时间一致。
【水分】:不得过%。
(库仑滴定)【比旋度】+~+。
供试品溶液:10 mg/ mL,溶于甲醇,20,以无水和无溶剂物计。
【炽灼残渣】不得过%。
【重金属】不得过20 ppm。
(重金属第二法)【有关物质】稀释液,溶液A,溶液B,峰鉴别溶液,色谱系统——同含量项下。
标准溶液——直接采用含量项下标准溶液。
供试品溶液——直接采用含量项下供试品溶液。
程序——分别进样等体积(约10 µL)标准溶液和供试品溶液,注入色谱仪,记录谱图,测定所有峰面积。
用下列公式计算卡培他滨中每个杂质的百分比:100(1/F)(C S / C U)(r I / r S) F——各个杂质的相对响应因子,见表1CS——标准溶液中卡培他滨标准品的浓度,mg/mlCU——供试品溶液中卡培他滨的浓度,mg/mlrI——供试品溶液中各个杂质的峰响应rS——标准溶液中卡培他滨的峰响应限度见表1。
表 1CompoundRelativeRetentionTimeRelativeResponseFactor (F)Limit(%) Capecitabine related compound ACapecitabine related compound B2,3-Di-O-acetyl-5-deoxy-5-fluorocytidine5-Deoxy-5-fluoro-N4-(2-methyl-1-butyloxycarbonyl)cytidine +5-Deoxy-5-fluoro-N4-(3-methyl-1-butyloxycarbonyl)cytidineCapecitabine—[1-[5-Deoxy-3-O-(5-deoxy--D-ribofuranosyl)--D-ribofuranosyl]-5-fluoro-2-oxo-1,2-dihydropyrimidin-4-yl]-carbamicacid pentyl ester[1-[5-Deoxy-2-O-(5-deoxy--D-ribofuranosyl)--D-ribofuranosyl]-5-fluoro-2-oxo-1,2-dihydropyrimidin-4-yl]-carbamicacid pentyl esterCapecitabine related compound C[1-[5-Deoxy-3-O-(5-deoxy--D-ribofuranosyl)--D-ribofuranosyl]-5-fluoro-2-oxo-1,2-dihydropyrimidin-4-yl]-carbamicacid pentyl ester2,3-Di-O-acetyl-5-deoxy-5-fluoro-N4-(pentyloxycarbonyl)cytidineIndividual unspecified impurity—Total unspecified impurities——Total impurities——【含量】稀释液——水:甲醇:乙腈=60:35:5稀醋酸——%醋酸水溶液(v/v)溶液A——稀醋酸:甲醇:乙腈=60:35:5溶液B——甲醇:稀醋酸:乙腈=80:15:5标准溶液——精密称取一定量USP卡培他滨对照品,溶于稀释液,超声,并用稀释液稀释以获得浓度约为mL的溶液。

vivi2010-10-02USP总目录:修订文件1 New Official Text,修订公告。
勘误表,临时修订声明,修)加快修订过程包括勘误表,临时修订声明(IRAS上刊部分刊出,勘误表,临时修订公告也会在订公告在USP网站上New Official Text PF出前言2front matter药典与处方集增补删减情况,审核人员,辅料收录情况3凡例药典,标题和修订1 2 药典地位和法律认可 3标准复合性专论和通则45 专论组成6 检验规范和检验方法7 测试结果8 术语和定义处方和配药 910 包装存储与标签4通则章节列表)一般检查和含量测定(章节编号小于1000检查和含量分析的一般要求检查和含量分析的仪器,微生物检查,生物检查和含量测定,化学检查和含量测定,物理检查和测定1000一般信息(章节号大于)5食物补充剂通则试剂(试剂,指示剂,溶液等)6.参考表7性状描述和溶解性查询表(按字母顺序)食品补充剂各论(字母顺序)8各论(辅料标准)9NF10 USP各论11术语附件:通则的章节中文目录(使用起来比较方便,直接找对应章节号即可)一、通用试验和检定(1)试验和检定的总要求1 注射剂11 参比标准物(2)试验和检定的装置16 自动分析方法21 测温仪31 容量装置,如容量瓶、移液管、滴定管,各种规格的误差限度41 砝码和天平(3)微生物学试验51 抗菌效力试验55 生物指示剂:耐受性能试验61 微生物限度试验61 非灭菌制品的微生物检查:计数试验62 非灭菌制品的特定菌检查,如大肠杆菌、金葡菌、沙门氏菌等71 无菌试验(4)生物学试验和检定81 抗生素微生物检定85 细菌内毒素试验87 体外生物反应性试验:检查合成橡胶、塑料、高聚物对哺乳类细胞培养的影响88 体内生物反应性试验:检查上述物质对小鼠、兔iv、ip或肌内植入的影响泛酸钙检定91111 生物检定法的设计和分析115 右泛醇检定121 胰岛素检定141 蛋白质——生物适应试验,用缺蛋白饲料大鼠,观察水解蛋白注射液和氨基酸混合物的作用151 热原检查法161 输血、输液器及类似医疗装置的内毒素、热原、无菌检查171 维生素B活性检定12(5)化学试验和检定A 鉴别试验181 有机含氮碱的鉴别191 一般鉴别试验193 四环素类鉴别197 分光光度法鉴别试验201 薄层色谱鉴别试验B 限量试验206 铝211 砷221 氯化物和硫酸盐223 二甲基苯胺226 4-差向脱水四环素231 重金属241 铁251 铅261 汞271 易炭化物试验281 炽灼残渣291 硒C 其他试验和检定中和酸能力301311 藻酸盐检定331 苯丙胺检定341 多剂量容器注射剂中所加防腐剂含量的气相色谱或极谱法测定345 枸橼酸与其盐以及磷酸盐检定351 甾体检定361 巴比妥酸盐检定371 维生素B放射示踪物检定12381 注射剂橡胶塞检查391 肾上腺素检定401 脂肪和固定油检查411 叶酸检定425 抗生素碘量法检定429 微粒大小的光衍射测量431 甲氧基测定441 烟酸或烟酰胺检定451 亚硝酸盐滴定461 氮测定466 普通杂质的薄层色谱法检查467 有机挥发性杂质检查法467 残留溶剂测定471 氧瓶燃烧法481 核黄素检定501 有机含氮碱的盐511 单一甾醇检定521 磺胺类的色谱法检定531 硫胺检定541 滴定法554 α-生育酚检定561 植物来源物品的一般检查项目植物来源物品的各种鉴别项目(植物学部分、显微鉴别、化学鉴别)563 565 植物提取物的一般提取方法和要求571 维生素A检定:化学法、色谱法581 维生素D检定:色谱法、化学法、生物法591 锌测定(6)物理试验和测定601 气雾剂、鼻喷雾剂、计量吸入剂和干粉吸入剂的各项检测611 乙醇含量测定:蒸馏法、气一液色谱法616 固体的疏松密度和叩击密度测定621 色谱法631 色度检查和标准641 溶解的完全性检查643 总有机炭测定645 水导电性测定651 冻凝温度的测定661 药用容器的检测项目要求671 盛装胶囊和片剂容器加盖后对湿气的通透性试验691 棉花吸附性和纤维长度测定695 结晶性检查696 用溶液测热法测定结晶度698 装量检查699 固体密度(粉粒密度测定法)701 崩解试验711 溶出试验721 蒸馏温度范围(馏程)测定724 通过透皮转运系统药物的释放726 电泳727 毛细管电泳730 等离子体光谱化学检查法731 干燥失重炽灼失重733736 质谱法741 熔点范围或温度的测定751 眼膏中的金属颗粒测定755 最低装量检查法761 核磁共振771 眼用软膏的要求776 光学显微镜微粒检查法781 旋光度检查785 渗透压摩尔浓度测定法786 用分析筛测量颗粒大小的分布788 注射液中微粒物质测定法789 眼用溶液中微粒物质测定法791 PH测定法795 非灭菌制剂的药物配制要求797 灭菌制剂的药物配制要求801 极谱法811 粉末细度测定821 放射活性药物823 正电子发射层析X线摄影(PET)所用放射性药物的配制831 折光指数测定841 比重测定846 粉末的比表面积测定851 分光光度法与光散射861 外科缝合线直径检查871 附有针的缝合线检查881 外科缝合线、纺织品与膜片的弹力强度检查891 热分析:温度变化、热解重量分析、易熔杂质分析等905 剂量单位的均匀性检查(含量均匀度、装量差异)911 黏度测定药品含水量的测定921941 结晶型药物的X线衍射分析二、通用资料1010 数据分析方法1015 诊断用放射药的自动合成装置1031 药用容器、医用装置和植入物所用材料的生物相容性检查1035 灭菌用生物指示剂1041 生物制品的批签发1043 细胞、基因和组织工程产品的辅助材料1045 生物技术产品1046 细胞和基因治疗产品1047 生物技术产品的检验法1048 生物技术产品的质量——重组DNA蛋白质产品生产所用细胞表达构成的分析1049 生物技术产品的稳定性试验1050 人或动物来源的细胞系所得生物技术产品的病毒安全性评价1051 玻璃仪器清洗方法1061 颜色的仪器测量1065 离子色谱1072 消毒剂与防腐剂1074 赋形剂生物学安全性评价指导原则1075 复方药物配制质量规范1078 大批量药用赋形剂的生产质量规范1079 储存与运输的质量规范1081 明胶的凝胶强度1086 药品中的杂质来源1087 特性溶出1088 剂型的体外和体内评价1090 体内生物等效性试验指导原则1091 剂型中含有无活性组分的标示1092 溶出试验方法的发展和验证药用滴管11011111 非灭菌药品的微生物特征1111 非灭菌药品的微生物特征检查:药用原料和药物制剂的判定标准1112 非灭菌药品中的水活性测定,即在同一温度时,药品中水的蒸气压与纯水蒸气压之比,它等于药品在密闭系统中产生相对湿度的1%1116 清洁室和其他受控环境的微生物评价1117 微生物实验室的质量规范(GLP)1118 监控装置:时间、温度、湿度1119 近红外分光光度法1120 拉曼(Raman)分光光度法1121 药品命名法1136 药品包装:应用单元1146 口服固体药分装在单疗程剂量容器中的检查方法1150 药物剂型的稳定性1151 药物剂型1160 处方调配的药学计算1171 原料药的位相溶解度分析1174 粉末流动性测定1176 处方天平和容量装置1177 包装质量规范1178 分装质量规范1181 扫描电子显微镜1191 调剂工作中的药品稳定性保持1196 药典协调(指欧洲药典、美国药典、日本药局方三方机构讨论协调的原则和方法)1207 灭菌产品包装:完整性评价1208 灭菌试验:隔离系统的验证1209 灭菌:化学和物理化学的指示剂与积分仪1211 药典收载品种的灭菌和灭菌保证1216 片剂脆性检查1221 茶匙(家用标准为5 ml,可作为病人口服液体药物的量具,误差应小于10%)药品灭菌终点的放行参数12221223 微生物替代方法的验证1225 药典方法的验证1227 在抗菌效力、微生物限度、灭菌等试验中,微生物的恢复验证1230 血液透析用水1231 药用水的制备和要求1241 在制药系统中,水—固体的相互作用1251 用分析天平称量的要求1265 书写药物处方的指导原则三、饮食增补剂2021 营养和饮食增补剂的微生物计数试验2022 营养和饮食增补剂中不允许存在的微生物(如金葡菌、沙门氏菌、大肠杆菌、梭状芽胞杆菌属)检查法2023 非灭菌的营养和饮食增补剂中的微生物特征2030 植物来源物品的增补资料2040 饮食增补剂的崩解和溶出检查2091 饮食增补剂的重(装)量差异检查2750 饮食增补剂的生产条件与质量要求(与药品有别)。
621CHROMATOGRAPHY色谱法INTRODUCTION介绍This chapter defines the terms and procedures used in chromatography and provides general information. Specific requirements for chromatographic procedures for drug substances and dosage forms, including adsorbent and developing solvents, are given in the individual monographs.此章节定义了色谱法中用到的术语和步骤,并提供了通用信息。
对于原料药和成药的色谱步骤的具体要求,包括吸附剂和展开溶剂,在具体各论中给出。
Chromatography is defined as a procedure by which solutes are separated by a dynamic differential migration process in a system consisting of two or more phases, one of which moves continuously in a given direction and in which the individual substances exhibit different mobilities by reason of differences in adsorption, partition, solubility, vapor pressure, molecular size, or ionic charge density. The individual substances thus separated can be identified or determined by analytical procedures.色谱法是应用溶质在两相或多相系统中的差速迁移来进行分离的技术,其中一相持续地向特定方向移动,而由于物质在吸附性、分配、溶解性、气体压力、分子大小、或离子电荷密度上的差异,会显示出不同的移动性。
氨苄西林钠Ampicillin Sodium(USP31-NF26第1420页)C16H18N3NaO4S 371.39按无水物计算,氨苄西林钠的效价以氨苄西林(C16H19N3O4S)计,每1mg 应为845µg~988µg。
[包装和贮存]贮存在密闭容器中。
[标贴] 本品用于制备注射剂时,标贴上应注明本品无菌或本品在制备注射剂过程中须经进一步的处理。
[USP标准物质] <11> USP氨苄西林RS、USP氨苄西林钠RS、USP内毒素RS。
[鉴别] A:红外光吸收图谱<179M>。
B:显钠盐反应<191>。
[结晶性] <695> 应符合要求。
(注-如为冻干粉则可不检此项)[pH] <791> 取本品,加水制成每1mL中含10.0mg氨苄西林的溶液,振摇使溶解,pH值应为8.0~10.0。
[水分] <921> 不得过2.0%。
方法Ⅰ。
[二甲基苯胺] <223> 应符合要求。
内标溶液、对照品溶液、供试品溶液的制备方法如下:内标溶液:取N,N-二乙基苯胺75mg,加1mol/L盐酸溶液使溶解,用水逐步稀释制成每1mL中约含30µg的溶液。
对照品溶液:取N,N-二甲基苯胺50.0mg,精密称定,置50mL量瓶中,加1mol/L盐酸溶液25mL振摇混匀后,用水稀释至刻度,摇匀,精密量取2mL,置100mL量瓶中,用水稀释至刻度,摇匀,精密量取1mL,置适宜的离心管中,精密加入1.25mol/L氢氧化钠溶液、内标溶液和环己烷各1mL,强烈振摇1分钟然后离心,取上清液,作为对照品溶液。
供试品溶液:取本品1.0g,精密称定,置适宜的离心管中,加1.25mol/L 氢氧化钠溶液2mL,振摇使溶解,精密加入内标溶液1mL和环己烷1mL,强烈振摇1分钟然后离心,取上清液,作为供试品溶液。
色谱系统(见色谱法<621>):气相色谱仪应配有火焰离子化检测器和2mm×2m的填充有上涂3%液相G3的硅烷化S1A的色谱柱。
美国药典USPNF色谱随着药物研究和生产的不断发展,化学分析成为药物质量控制的重要手段之一。
毒理学和药物安全性评估需要准确的组成分析和含量测定,而且质量控制必须遵循有国际影响力的标准。
美国药典(USP)是一个评估和监管药物质量的非营利组织,在美国以及全球范围内都被广泛认可,它为全球药物和食品行业的质量标准制定提供了重要的参考依据。
其中与色谱分析相关的内容被编入美国药典的色谱单元,成为美国药典色谱(USP-NF)。
美国药典色谱(USPNF)是美国药典所编制的、与色谱分析相关的内容所组成的分册。
该分册覆盖了各种技术方法和分析技巧,在色谱学分析方面,主要包含了高效液相色谱(HPLC)和气相色谱(GC)两类。
HPLC作为一种重要的分离方法,广泛应用于药物分析、生化分析、食品分析等领域。
美国药典色谱通过列出HPLC检测专用的药物物质,以及制定适宜的HPLC方法进行药物物质分析,以保障药品质量。
美国药典色谱还包含针对气相色谱分析方法的指南。
GC作为一种分离技术,已经成为许多行业中最常用的分析方法之一。
通过美国药典颁布的气相色谱方法,可以保证药品的质量,满足全球质量标准,在药物的生产和分析中发挥着重要的作用。
除此之外,USPNF还包含了一些在特殊情况下使用的其他色谱方法,如离子交换色谱、薄层色谱等。
在它们列举的分析方法中,可以为药品分析提供多种参考依据,同时保障质量的一致性和准确性。
美国药典色谱(USPNF)不仅仅是一个规范和标准操作指南,它还为药物制造商提供了一个国际公认的质量控制体系。
在全球化的药品市场中,美国药典色谱既刻画了一种药品质量目标,也规定了药品质量标准,具有非常重要的临床和药学意义。
同时,由于美国药典色谱列举的方法都是公认的正规方法,因此在使用美国药典列举的分析方法时,药品制造商可以对质量及其相应的操作有更好的把控,同时也为药品设计提供更多参考依据。
总之,美国药典色谱作为美国药典中的一个重要分册,可以为全球药品和食品行业提供良好的质量和标准化分析方法,保障药品和食品的安全性和质量一致性。
Saccharin SodiumC 7H 4NNaO 3S·2H 2O 241.201,2-Benzisothiazol-3(2H )-one, 1,1-dioxide, sodium salt, dihydrate.1,2-Benzisothiazolin-3-one 1,1-dioxide sodium salt dihydrate [6155-57-3].Anhydrous 205.17[128-44-9].» Saccharin Sodium contains not less than 99.0 percent and not more than 101.0 percent of C 7H 4NNaO 3S·2H 2O, calculated on the anhydrous basis. Packaging and storage— Preserve in well-closed containers. Store at roomtemperature.Labeling— Where the quantity of saccharin sodium is indicated in the labeling of any preparation containing Saccharin Sodium, this shall be expressed in terms of saccharin (C 7H 5NO 3S).USP Reference standards 11— USP Saccharin Sodium RS .USP o-Toluenesulfonamide RS .USP p-Toluenesulfonamide RS .Clarity of solution— [NOTE—The Test solution is to be compared to Referencesuspension A and to water in diffused daylight 5 minutes after preparation of Reference suspension A.]Hydrazine solution— Transfer 1.0 g of hydrazine sulfate to a 100-mL volumetric flask, dissolve in and dilute with water to volume, and mix. Allow to stand for 4 to 6 hours. Methenamine solution— Transfer 2.5 g of methenamine to a 100-mL glass-stoppered flask, add 25.0 mL of water, insert the glass stopper, and mix to dissolve.Primary opalescent suspension— [NOTE—This suspension is stable for 2 months,provided it is stored in a glass container free from surface defects. The suspension must not adhere to the glass and must be well mixed before use.] Transfer 25.0 mL ofHydrazine solution to the Methenamine solution in the 100-mL glass-stoppered flask. Mix, and allow to stand for 24 hours.Opalescence standard— [NOTE—This suspension should not be used beyond 24 hoursafter preparation.] Transfer 15.0 mL of the Primary opalescent suspension to a 1000-mL volumetric flask, dilute with water to volume, and mix.Reference suspensions— Transfer 5.0 mL of the Opalescence standard to a 100-mL volumetric flask, dilute with water to volume, and mix to obtain Reference suspension A. Transfer 10.0 mL of the Opalescence standard to a second 100-mL volumetric flask, dilute with water to volume, and mix to obtain Reference suspension B.Test solution— Dissolve 5.0 g of test material in about 20 mL of a 200 g per L solution of sodium acetate, dilute with the same solution to 25 mL, and mix.Procedure— Transfer a sufficient portion of the Test solution to a test tube of colorless, transparent, neutral glass with a flat base and an internal diameter of 15 mm to 25 mm to obtain a depth of 40 mm. Similarly transfer portions of Reference suspension A, Reference suspension B, water, and a 200 g per L solution of sodium acetate to separate matching test tubes. Compare the Test solution, Reference suspension A, Reference suspension B, water, and a 200 g per L solution of sodium acetate in diffused daylight, viewing vertically against a black background (see Visual Comparison underSpectrophotometry and Light-Scattering 851). [NOTE—The diffusion of light must be such that Reference suspension A can readily be distinguished from water, and that Reference suspension B can readily be distinguished from Reference suspension A.] The Test solution shows the same clarity as that of water, or the 200 g per L solution of sodium acetate, or its opalescence is not more pronounced that that of Reference suspension A.Color of solution—Standard stock solution— Combine 3.0 mL of ferric chloride CS, 3.0 mL of cobaltous chloride CS, 2.4 mL of cupric sulfate CS, and 1.6 mL of dilute hydrochloric acid (10 g per L).Standard solution— [NOTE—Prepare the Standard solution immediately before use.] Transfer 1.0 mL of the Standard stock solution to a 100-mL volumetric flask, dilute with dilute hydrochloric acid (10 g per L) to volume, and mix.Test solution— Use the Test solution from the test for Clarity of solution.Procedure— Transfer a sufficient portion of the Test solution to a test tube of colorless, transparent, neutral glass with a flat base and an internal diameter of 15 mm to 25 mm to obtain a depth of 40 mm. Similarly transfer portions of the Standard solution, a 200 g per L solution of sodium acetate, and water to separate matching test tubes. Compare the Test solution, the Standard solution, a 200 g per L solution of sodium acetate, and water in diffused daylight, viewing vertically against a white background (see Visual Comparisonunder Spectrophotometry and Light-Scattering 851). The Test solution has the appearance of water or of the 200 g per L solution of sodium acetate, or is not moreintensely colored than the Standard solution.Identification—A: Infrared Absorption 197K —Test specimen— Dry the specimen at 105 to constant weight.B: To a solution (1 in 10) add 2 mL of 15% potassium carbonate, and heat to boiling. No precipitate is formed. Add 4 mL of Potassium pyroantimonate solution, and heat to boiling. Allow to cool in ice water and, if necessary, rub the inside of the test tube with a glass rod.A dense precipitate is formed.Potassium pyroantimonate solution—Dissolve 2 g of potassium pyroantimonate in 95 mL of hot water. Cool quickly, and add a solution containing 2.5 g of potassium hydroxide in 50 mL of water and 1 mL of sodium hydroxide solution (8.5 in 100). Allow to stand for 24 hours, filter, and dilute with water to 150 mL.C: Sodium salts impart an intense yellow color to a nonluminous flame.Acidity or alkalinity— To a solution of 1.0 g in 10 mL of carbon dioxide-free water add 1 drop of phenolphthalein TS: no pink color is produced. Then add 1 drop of 0.1 N sodium hydroxide: a pink color is produced.Water, Method I 921: not more than 15.0%. Readily carbonizable substances 271— Dissolve 200 mg in 5 mL of sulfuric acid(between 94.5% and 95.5% [w/w] of H 2SO 4), and maintain at a temperature of 48 to 50 for 10 minutes: the solution has no more color than Matching Fluid A, when viewed against a white background.Heavy metals, Method I 231— Dissolve 4 g in 46 mL of water, add 4 mL of dilute hydrochloric acid (1 in 12), mix, and rub the inner wall of the vessel with a glass rod until crystallization begins. Allow the solution to stand for 1 hour, then pass through a dry filter, discarding the first 10 mL of the filtrate, and use 25 mL of the subsequent filtrate for the Test Preparation: the limit is 0.001%.Limit of toluenesulfonamides—Internal standard solution— Dissolve 25 mg of caffeine in methylene chloride, and dilute with the same solvent to 100 mL.Reference solution— Dissolve 20.0 mg of USP o -Toluenesulfonamide RS and 20.0 mg of USP p -Toluenesulfonamide RS in methylene chloride, and dilute with the same solvent to 100.0 mL. Dilute 5.0 mL of the solution with methylene chloride to 50.0 mL. Evaporate 5.0 mL of the final solution to dryness in a stream of nitrogen. Dissolve the residue in 1.0 mL of the Internal standard solution.Test solution— Dissolve 10.0 g of the substance to be examined in about 45 mL of water. If necessary, adjust the solution with 1 N sodium hydroxide or 1 N hydrochloric acid to a pH of 7 to 8, and dilute with water to 50 mL. Shake the solution with four quantities each of 50 mL of methylene chloride. Combine the lower layers, dry over anhydrous sodium sulfate, and filter. Wash the filter and the sodium sulfate with 10 mL of methylene chloride. Combine the solution and the washings, and evaporate almost to dryness in a water bathat a temperature not exceeding 40. Using a small quantity of methylene chloride, quantitatively transfer the residue into a suitable 10-mL tube, evaporate to dryness in a stream of nitrogen, and dissolve the residue in 1.0 mL of the Internal standard solution.Blank solution— Evaporate 200 mL of methylene chloride to dryness in a water bath at atemperature not exceeding 40. Dissolve the residue in 1 mL of methylene chloride.Chromatographic system (see Chromatography 621)— The gas chromatograph is equipped with a flame-ionization detector and contains a 0.53-mm × 10-m fused silica column, coated with a 2-µm thickness of phase G3. The injection port, column, and detector temperatures are maintained at about 250, 180, and 250, respectively; and nitrogen is used as the carrier gas at a flow rate of about 10 mL per minute. The injector employs a split ratio of 1:2.Procedure— Inject about 1 µL of the Reference solution. Adjust the sensitivity of the detector so that the height of the peak due to caffeine is not less than 50% of the full scale of the recorder. The substances are eluted in the following order: o-toluenesulfonamide,p-toluenesulfonamide, and caffeine. The test is not valid unless the resolution between the peaks due to o-toluenesulfonamide and p-toluenesulfonamide is at least 1.5. Inject about 1 µL of the Blank solution. In the chromatogram obtained, verify that there are no peaks with the same retention times as the internal standard, o-toluenesulfonamide, and p-toluenesulfonamide. Inject about 1 µL of the Test solution and 1 µL of the Reference solution. If any peaks due to o-toluenesulfonamide and p-toluenesulfonamide appear in the chromatogram obtained with the Test solution, the ratio of their areas to that of the internal standard is not greater than the corresponding ratio in the chromatogram obtained with the Reference solution (10 ppm of o-toluenesulfonamide and 10 ppm of p-toluenesulfonamide).Limit of benzoate and salicylate— To 10 mL of a solution (1 in 20), previously acidified with 5 drops of 6 N acetic acid, add 3 drops of ferric chloride TS: no precipitate or violet color appears.Organic volatile impurities, Method IV 467: meets the requirements.(Official until July 1, 2008)Assay— Dissolve, with the aid of slight heating if necessary, about 150 mg of Saccharin Sodium, accurately weighed, in 50 mL of glacial acetic acid. Titrate with 0.1 N perchloricacid, determining the endpoint potentiometrically. Perform a blank titration, if necessary, and make the appropriate correction. Each mL of 0.1 N perchloric acid is equivalent to 20.52 mg of C 7H 4NNaO 3S.Auxiliary Information— Before contacting USP, have you checked for your question in the FAQs ?USP31–NF26 Page 3208Pharmacopeial Forum : Volume No. 32(4) Page 1114Chromatographic Column— SACCHARIN SODIUMChromatographic columns text is not derived from, and not part of, USP 31 or NF 26. Topic/Question Contact Expert Committee Monograph Kevin T. Moore, Ph.D.Scientist1-301-816-8369(EM105) Excipient Monographs 1Reference StandardsLili Wang, Technical ServicesScientist1-301-816-8129RSTech@。