肌张力障碍临床诊断治疗指南
- 格式:doc
- 大小:14.65 KB
- 文档页数:5


肌张力障碍锁定本词条由好大夫在线提供内容并参与编辑。
王玉平(主任医师)首都医科大学宣武医院神经内科卫华(副主任医师)首都医科大学宣武医院神经内科肌张力障碍(dystonia)是主动肌与拮抗肌收缩不协调或过度收缩引起的以肌张力异常的动作和姿势为特征的运动障碍综合征,具有不自主性和持续性的特点。
依据病因可分为原发性和继发性。
原发性肌张力障碍与遗传有关。
继发性肌张力障碍包括一大组疾病,有的是遗传性疾病(如肝豆状核变性,亨廷顿舞蹈病,神经节苷脂病等),有的是由外源性因素引起的(如围生期损伤、感染、神经安定药物)。
西医学名肌张力障碍英文名称dystonia 所属科室内科- 神经内科主要病因遗传因素目录1 疾病分类2 病因及发病机制3 临床表现▪扭转痉挛▪痉挛性斜颈▪Meige综合征▪手足徐动症▪书写痉挛4 诊断及鉴别诊断▪诊断▪鉴别诊断5 疾病治疗▪药物治疗▪注射A型肉毒毒素▪手术6 疾病预后7 疾病预防疾病分类依据肌张力障碍的发生部位,可分为局限性、节段性、偏身性和全身性。
一般而言,发病年龄越早,症状可能越严重,波及身体其他部位的可能性也越大。
发病年龄越大,肌张力障碍越可能保持其局灶性。
局限性肌张力障碍指肌张力障碍只影响到躯体的一部分,如痉挛性斜颈、书写痉挛、眼睑痉挛、口-下颌肌张力障碍等。
节段性肌张力障碍累及一个以上相邻部位,如Meige综合征(眼、口和下颌),一侧上肢加颈部,双侧下肢等。
累及一侧身体时称偏侧肌张力障碍,一般由对侧大脑半球病变所致。
全身性肌张力障碍,累及至少一个节段,加上一个以上其他部位。
病因及发病机制原发性肌张力障碍多为散发,少数有家族史,呈常染色体显性或隐性遗传,或X染色体连锁遗传,最多见于7~15岁儿童或少年。
常染色体显性遗传的原发性扭转痉挛绝大部分是由定位在9q32-34的DYTl基因突变所致,外显率为30%~50%。
多巴反应性肌张力障碍也是常染色体显性遗传,为三磷酸鸟苷环化水解酶-1(GCH-1)基因突变所致。


肌张力障碍诊断与治疗指南肌张力障碍(TBD)是一组神经系统疾病,主要特征为肌肉过度紧张、抽搐和不协调运动。
肌张力障碍包括多种类型,如帕金森病、帕金森综合症、帕金森样综合症、震颤麻痹综合症等。
肌张力障碍的诊断和治疗需要综合考虑患者的症状、体征、血液检测结果以及相关影像学检查。
下面将介绍肌张力障碍的诊断和治疗指南。
一、诊断指南:1.病史询问:详细了解患者的临床表现和症状变化情况,包括运动障碍、不自主运动、肌肉僵硬等。
2.体格检查:检查患者的神经系统、肌肉和运动功能,观察是否存在震颤、肌张力增高、运动不协调等症状。
3.神经影像学检查:如头颅CT、MRI等,可以帮助鉴别肌张力障碍和其他神经系统疾病。
4. 血液检测:可以测定血清中特定标志物的水平,如帕金森病的标志物为α-synuclein。
还可以检测其他相关指标,如血清铜蓝蛋白等。
5.脑电图(EEG):可以观察脑电波的异常变化,帮助判断肌张力障碍的类型和严重程度。
6.遗传学检测:对于早发性肌张力障碍,一些家族性和遗传性因素可能起到关键作用,因此遗传学检测可以帮助确定诊断。
二、治疗指南:1.药物治疗:药物治疗是肌张力障碍的主要治疗方法,不同类型的肌张力障碍可能需要使用不同的药物。
常用的药物包括抗帕金森病药物、抗震颤药物、抗抽搐药物等。
治疗过程中需要根据患者的症状和体征来调整药物剂量和种类。
2.物理治疗:物理治疗可以通过肌肉放松、增强肌力、改善运动协调性等方式来缓解肌张力障碍的症状。
常见的物理治疗方法包括按摩、热敷、理疗、康复训练等。
3.手术治疗:对于一些严重的肌张力障碍患者,药物和物理治疗效果不佳时,可以考虑手术治疗。
常见的手术方式包括脑深部电刺激(DBS)和肌肉松弛剂注射,手术治疗需要慎重选择,风险与利益需要充分评估。
总结:肌张力障碍的诊断和治疗需要综合考虑患者的临床症状和体征、影像学检查结果以及血液检测结果。
药物治疗是主要的治疗方式,物理治疗和手术治疗可作为辅助。

中国重症肌无力诊断和治疗指南一、本文概述《中国重症肌无力诊断和治疗指南》是一份权威的医学指南,旨在为医生和医疗工作者提供关于重症肌无力(Myasthenia Gravis, MG)的标准化诊断和治疗建议。
重症肌无力是一种由自身免疫系统异常引起的神经-肌肉接头传递障碍性疾病,临床表现为部分或全身骨骼肌无力和易疲劳。
本指南的编写目的是确保患者能够得到及时、准确和个性化的医疗服务,改善其生活质量,降低疾病对社会和家庭的影响。
本指南涵盖了重症肌无力的流行病学、发病机制、临床表现、诊断方法、治疗策略以及疾病管理等方面的内容。
通过全面系统地介绍重症肌无力的最新研究进展和临床实践经验,本指南旨在为医生提供科学、规范、实用的治疗建议,以促进重症肌无力患者的康复。
本指南也为政策制定者、医学教育工作者和科研人员提供了有价值的参考信息,有助于推动我国重症肌无力诊疗水平的提高。
本指南的制定遵循了科学、规范、实用的原则,充分借鉴了国内外相关指南和研究成果,结合我国实际情况,形成了具有中国特色的重症肌无力诊断和治疗方案。
我们希望本指南能够为广大医生和医疗工作者提供有益的帮助,共同推动我国重症肌无力诊疗事业的发展。
二、重症肌无力的病因与病理生理机制重症肌无力(Myasthenia Gravis,MG)是一种慢性自身免疫性疾病,主要表现为神经-肌肉接头的传递功能障碍。
其病因复杂,涉及遗传、免疫、环境等多因素。
遗传因素在MG的发病中起着重要作用,许多研究已经证实,MG与主要组织相容性复合体(MHC)基因、乙酰胆碱受体(AChR)基因、补体基因、T细胞受体基因、免疫球蛋白基因以及细胞因子基因等相关。
在病理生理机制方面,MG主要是由于患者体内产生了针对乙酰胆碱受体(AChR)的自身抗体,导致神经-肌肉接头的突触后膜乙酰胆碱受体数量减少或功能受损,进而影响了神经冲动的传递,使骨骼肌发生易疲劳现象。
MG患者体内还可能存在针对肌肉特异性激酶(MuSK)和低密度脂蛋白受体相关蛋白4(LRP4)的自身抗体,这些抗体同样可以影响神经-肌肉接头的功能。

肌张力障碍病历模板病例标题:肌张力障碍病历患者信息:姓名: [患者姓名]性别: [患者性别]年龄: [患者年龄]联系方式: [患者联系方式]主诉: [患者主诉]现病史:1. [病情开始时间]2. [症状描述]3. [病情发展过程]既往史:1. [手术史]2. [药物使用史]3. [过敏史]4. [其他疾病史]家族史:1. [家族成员是否有肌张力障碍史]个人史:1. [吸烟史]2. [饮酒史]3. [饮食习惯]4. [体育锻炼情况]体格检查:1. 一般情况: [患者意识、精神状态]2. 神经系统: [肌张力状况、肢体活动度]3. 感觉系统: [触觉、温度、疼痛等感觉检查结果]4. 皮肤: [皮肤情况、皮肤病变情况]5. 其他系统检查: [如必要,根据具体情况填写其他系统检查结果]辅助检查:1. [血常规]2. [肌电图]3. [脑电图]4. [头颅磁共振成像(MRI)]5. [其他辅助检查]诊断、治疗计划与过程:1. 临床诊断: [根据患者症状和体格检查结果提供初步诊断]2. 确诊依据: [辅助检查结果]3. 计划与过程: [治疗计划和过程,注明使用的药物、剂量和疗程]4. 治疗效果: [患者在治疗过程中的症状改善情况,如有需要可提供疗效评估指标]随访与复诊:1. 随访时间: [规定的随访时间]2. 随访结果: [患者随访的结果,症状变化等]3. 复诊建议: [根据患者随访结果,提供进一步治疗或复诊建议]医生签名: [医生签名]日期: [填写日期]。

肌张力障碍由于肌张力的改变引起的持续的异常的姿势与正在进行中的动作的阻断.分类肌张力障碍可分为全身性,局灶性或节段性.西医定义肌张力障碍是唯一或主要以颈、躯干及四肢的近端肌肉缓慢、持续、强烈扭转样不自主运动为表现的一种锥体外系疾患。
肌张力障碍的概念较混乱,症状及分类较复杂。
通常按病因可分为特发性与症状性肌张力障碍二类;按种族分为犹太及非犹太族二类;根据遗传学特点可分为常染色体隐性遗传、常染色体显性遗传及散发型三类;根据临床表现可分为典型,局限型及变异型三大类。
疾病症状典型的特发性肌张力障碍病例常以肢体的某一部分出现不自主运动起病,如通常以一侧足的不自主跖屈及内翻开始,先后扩展到同侧及对侧肢体,发生不自主扭曲运动;也有部分病例以头及手臂首先起病,在打字或写字时出现手的过度屈曲似书写痉挛;或颈部不自主扭转似痉挛性斜颈,早期仅在情绪激动或快步行走时,偶尔出现肢体的扭曲运动。
逐渐在走路、站立时也可出现这种异常不自主运动。
肌张力障碍运动发作是缓慢的、不随意和无目的的四肢及躯干的强烈扭转运动,往往造成步行缓慢,步履困难;如果面、颈肌受累可造成讲话不清。
随后,即使安静休息时扭转痉挛状态仍持续存在,通常表现为踝关节跖屈、旋转,膝关节伸展或屈曲,髋关节轻度屈曲,肘关节伸展,前臂内旋,拇指旋外屈曲。
由于受累肌肉的肌张力显著增高,因此这样的扭转痉挛姿势很难被意志控制或被动地纠正,仅在睡眠中方消失。
晚期,由于肌腱挛缩及肌肉纤维化引致固定畸形,虽睡眠中也持续存在。
本病患者的肌力并不减弱,但往往因不自主运动或肌肉孪缩而影响随意运动。
深反射无改变,无病理反射,深浅感觉正常。
常可因不自主运动引起肌肉肥大,但晚期往往出现废用性萎缩。
部分病人,末期可能有智能减退。
Cooper将本病分为以下5期。
第1期:往往在精神应激或随意运动引致某一个姿势时才出现肌张力障碍运动。
第2期:在随意运动过程中,肌张力障碍姿势持续存在,但安静时不出现。
第3期:安静时肌张力障碍姿势也持续存在,且主动或被动地纠正较困难。


临床肌张力障碍病因、临床表型及诊断要点肌张力障碍定义及流行病学肌张力障碍是一种以持续性或间歇性肌肉收缩导致重复运动和/或姿势异常为特征的运动障碍。
肌张力障碍性运动通常具有模式化的扭转型,可伴有震颤,常由随意运动引发或加重,且和肌肉兴奋的泛化相关。
肌张力障碍可单独存在,亦可与其他运动障碍疾病表型及神经系统疾病特征同时存在,是第三大常见的运动障碍。
肌张力障碍多数情况下持续存在,亦可为任务特异性、阵发性及日间性。
肌张力障碍是一组不同的疾病,临床异质性大,患病率亦各异,其中累及身体单个部位的原发性局灶型肌张力障碍(以颈肌张力障碍最常见)比全身型肌张力障碍更为常见,患病率为16.4/100,000。
肌张力障碍的患病率随着年龄增长而增加,女性高于男性(尤其是局灶型和节段型肌张力障碍),且有种族、民族和地理差异。
肌张力障碍的分类主要依据临床特征和病因学两个维度,临床特征包括发病年龄、身体分布、时间模式和伴随特征(合并其他运动障碍或神经体征);病因学包括神经系统病理和遗传。
局灶型肌张力障碍及临床特征颈肌张力障碍(Cervical dystonia)曾称为“痉挛性斜颈”,是最常见的特发性肌张力障碍,常中年发病,多以颈部疼痛起病。
该病可累及颈部的深、浅肌肉,但以胸锁乳突肌、斜方肌及颈夹肌的收缩最常见,患者可能表现为异常的头部水平旋转(斜颈)、颈部侧倾、头部前屈(垂颈)、头部后伸(颈后倾)或组合症状。
30 - 60%患者伴有头部震颤,25%患者可伴有手部震颤(可模拟特发性震颤),通常有存在某种感觉诡计可缓解症状眼睑痉挛(Blepharospasm)好发于40 - 60 岁人群,女性多见,累及眼轮匝肌和其他眼周肌肉(含降眉间肌和皱眉肌),表现为眼轮匝肌和其他眼周肌肉的不自主收缩,导致间歇或持久性不自主瞬目,通常表现为双侧、同步对称,但也可不对称。
在注视、强光及紧张焦虑时加重。
可有长期性痉挛和功能性失明,可与下面部痉挛同时存在时(Meige 综合征)。

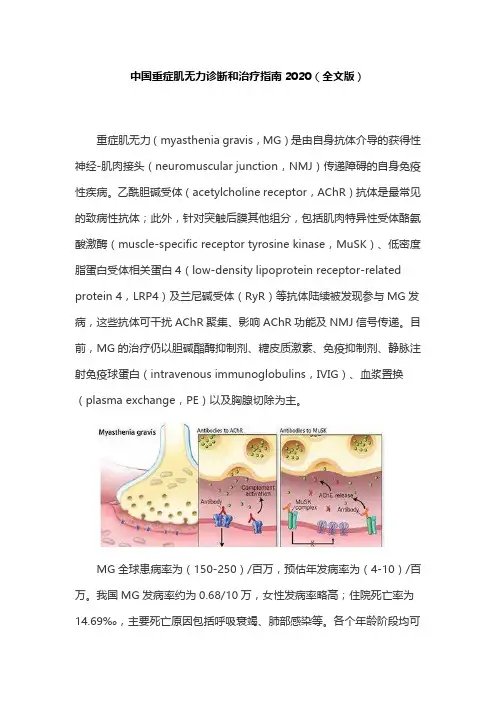
中国重症肌无力诊断和治疗指南2020(全文版)重症肌无力(myasthenia gravis,MG)是由自身抗体介导的获得性神经-肌肉接头(neuromuscular junction,NMJ)传递障碍的自身免疫性疾病。
乙酰胆碱受体(acetylcholine receptor,AChR)抗体是最常见的致病性抗体;此外,针对突触后膜其他组分,包括肌肉特异性受体酪氨酸激酶(muscle-specific receptor tyrosine kinase,MuSK)、低密度脂蛋白受体相关蛋白4(low-density lipoprotein receptor-related protein 4,LRP4)及兰尼碱受体(RyR)等抗体陆续被发现参与MG发病,这些抗体可干扰AChR聚集、影响AChR功能及NMJ信号传递。
目前,MG的治疗仍以胆碱酯酶抑制剂、糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白(intravenous immunoglobulins,IVIG)、血浆置换(plasma exchange,PE)以及胸腺切除为主。
MG全球患病率为(150-250)/百万,预估年发病率为(4-10)/百万。
我国MG发病率约为0.68/10万,女性发病率略高;住院死亡率为14.69‰,主要死亡原因包括呼吸衰竭、肺部感染等。
各个年龄阶段均可发病,30岁和50岁左右呈现发病双峰,中国儿童及青少年MG(juvenile myasthenia gravis,JMG)患病高达50%,构成第3个发病高峰;JMG 以眼肌型为主,很少向全身型转化。
最新流行病学调查显示,我国70-74岁年龄组为高发人群。
近年来,在MG诊疗方面取得了众多进展,积累了更多循证医学证据。
为此,中国免疫学会神经免疫分会基于近5年国内外文献中的最新证据,参考相关国际指南,反复讨论,在对中国MG诊治指南(2015)更新修订的基础上编写了本指南。
新指南采用MGFA临床分型替代Osserman 分型,旨在对疾病严重程度进行量化评估;提出MG亚组分类,指导精准化治疗;对治疗目标进行了定义;针对胸腺切除,利妥昔单抗、依库珠单抗等生物制剂的应用,眼肌型MG(ocular MG,OMG)早期免疫抑制治疗以及免疫检查点抑制剂(immune check point inhibitors,ICIs)治疗相关MG等方面提出了新的建议。
肌张力障碍诊疗规范2023版肌张力障碍(dystonia)是一种以持续性或间歇性肌肉收缩导致重复运动、姿势异常或二者兼有为特征的运动障碍病。
肌张力障碍性运动通常具有模式化、有扭转动作的特征,可伴有震颤,经常由主动运动启动或加重。
【病因与发病机制】肌张力障碍的病因和发病机制尚不明确。
其中遗传性肌张力障碍由基因突变所致。
自1991年OZeIiUS等在9号染色体发现了第f致病基因(z»m)以来,至少已有28个致病基因被定位。
【临床分类】肌张力障碍有许多分类方法,目前最新的由国际专家共识委员会修订的分类方法(2013)从两条主线(临床特征和病因学)进行了阐述,参见表23-6-4-1。
表23-6∙4-1肌张力障碍的分类按临床特征分类按病因学分类1.按肌张力隙碍本身的临床 1.按神经病理学改变分特征分(1)有神经系统退行性变(1)按起病年龄分的证据1)婴儿型:出生至2岁(2)有结构性病变的证据2)儿童型:3~12岁(3)无神经系统退行性3)少年型:13~20岁变/结构性病变的4)成人早期型:21~40岁证据5)成人晚期型:>40岁 2.按遗传性或获得性分(2)按肌张力障碍范围分(1)遗传性1)局灶性肌张力隙碍1)常染色体显性遗传2)节段性肌张力障碍2)常染色体隐性遗传3)多灶性肌张力隙碍3)X-性连锁隐性遗传4)偏身肌张力障碍4)线粒体遗传5)全身性肌张力隙碍(2)获得性(3)按病程分1)围产期脑损伤1)稳定型2)感染性疾病2)进展型3)药源性(4)按变异性分4)中毒性1)持续性5)血管性2)活动特异性6)肿瘤性3)日发性(白天)7)脑外伤4)发作性8)精神心理性2.按肌张力障碍相关的特(3)原发性征分1)散发性(1)肌张力障碍伴/或不伴其他运动2)家族性障碍1)单纯肌张力障碍2)联合性肌张力障碍(肌张力障碍联合其他运动障碍)(2)肌张力障碍合并其他神经系统或系统性疾病的表现【病理】原发性扭转痉挛可见非特异性的病理改变,包括壳核、丘脑及尾状核的小神经元变性消失,基底节的脂质及脂色素增多等。
肌张力障碍有哪些症状?*导读:本文向您详细介绍肌张力障碍症状,尤其是肌张力障碍的早期症状,肌张力障碍有什么表现?得了肌张力障碍会怎样?以及肌张力障碍有哪些并发病症,肌张力障碍还会引起哪些疾病等方面内容。
……*肌张力障碍常见症状:发作性睡眠、儿童型肌张力障碍、构音障碍、红细胞增多*一、症状肌张力障碍的临床分类目前尚不统一。
肌张力障碍的各种分类与临床特点相关,主要根据肌张力障碍的受累肢体和部位,可能造成肌张力障碍的原因,发病时的年龄等。
1.国内王维治主编的《神经病学》(2006年第1版)介绍如下分类(1)按病变累及部位分类:①局限性肌张力障碍:影响躯体某部,如痉挛性斜颈、痉挛性发音困难(声带)、睑痉挛、口-下颌肌张力障碍、书写痉挛(一侧上肢)、职业性痉挛、足肌张力障碍等。
②节段性肌张力障碍:表现节段性分布特点,如颅-颈部肌张力障碍,上肢伴或不伴中轴、头颈部肌张力障碍,下肢伴或不伴躯干肌张力障碍,躯干-颈部(不影响头面部)肌张力障碍等。
③多部位肌张力障碍:累及身体2个以上不靠近部位。
④广泛性肌张力障碍:影响广泛的躯体范围。
⑤偏侧性肌张力障碍:仅累及单侧身体。
(2)按病因分类:①遗传性进行性肌张力障碍:多在小儿发病,表现步行障碍及运动减少,躯干姿势异常,腰部明显向前弯曲,日内症状波动明显,晨轻,午后至傍晚加重,呈进行性发展,L-dopa疗效显著,本病与Parkinson病可能有类似之处。
②症状性肌张力障碍:如脑性瘫痪、累及基底核的脑卒中、Wilson病、Hallervorden-Spatz病、脑炎、脑肿瘤、精神病药物副反应等伴肌张力障碍,Parkinson病有时可伴肌张力障碍。
2.上海史玉泉等主编的《实用神经病学》(2004年第3版)则主张以下分类(1)按肌张力障碍范围的分类:①局限性肌张力障碍(focal dystonia):仅累及眼睑部肌群者,称眼睑痉挛(blepharospasm);累及口周及下颌肌群者,则称为口下颌肌张力障碍(oromandibular dystonia);疾病累及喉部肌群称为痉挛性构音障碍(spasmodic dysphonia);疾病累及颈部肌群称为痉挛性斜颈(spasmodic torticollis);疾病累及前臂和手部,称为书写性痉挛症(writer cramp)。
肌张力障碍诊断中国专家共识(2020年)1911年Oppenheimsh首次提出变形性肌张力障碍一词,描述具有异常运动、姿势扭转及肌张力波动变化的一种疾病[1]。
1984年国际肌张力障碍医学研究基金会顾问委员会提出了肌张力障碍的定义:一种不自主、持续性肌肉收缩引起的扭曲、重复运动或姿势异常的综合征[2]。
这个定义在相当长的时间里被临床医生和相关领域的研究者普遍接受并采用。
2008年中华医学会神经病学分会帕金森病及运动障碍学组制定了我国首个肌张力障碍诊断和治疗指南,对于肌张力障碍领域的规范诊断和治疗发挥了积极的作用[3]。
近10年来,国内外在肌张力障碍领域的研究非常活跃,特别是在遗传学方面取得重大进展[4-5],以脑深部电刺激为代表的治疗方法在疑难病例的治疗上获得突破[6-7],对于肌张力障碍的定义、分类、诊断、评价等方面也有了新的认识,因此在总结国内外最新研究成果的基础上,结合专家组的临床经验,制定了《肌张力障碍诊断中国专家共识》。
一、肌张力障碍的定义肌张力障碍是一种运动障碍,其特征是持续性或间歇性肌肉收缩引起的异常运动和(或)姿势,常重复出现。
肌张力障碍性运动一般为模式化的扭曲动作,可以呈震颤样。
肌张力障碍常因随意动作诱发或加重,伴有肌肉兴奋的泛化[8]。
“肌张力障碍”可用于描述一种具有独特表现的不自主运动,与震颤、舞蹈、抽动、肌阵挛等类同;也可用于命名一种独立的疾病或综合征,其中肌张力障碍症状是唯一或主要的临床表现,肌张力障碍是神经系统运动增多类疾病的常见类型。
肌张力障碍作为不自主运动的一种形式,常可以观察到以下现象[9]。
1.缓解技巧/感觉诡计(alleviating maneuvers/sensory tricks/gestesantagonistes):用于纠正异常姿势或缓解肌张力障碍性运动的随意动作,通常是涉及受累部位的简单运动,而不是用力对抗肌张力障碍症状。
2.镜像肌张力障碍(mirror dystonia):一种对侧运动诱发的单侧肢体的姿势或运动,与肌张力障碍的特征相同或类似,常见于受累较严重的一侧肢体。
肌张力障碍临床诊断治疗指南
【概述】
肌张力障碍(dystonia)是主动肌与拮抗肌收缩不协调或过
度收缩引起的以肌张力异常动作和姿势为特征的运动障碍
性疾病。
依据病因分为特发性和继发性;根据肌张力障碍的部位分为全身性、局限性、节段性、偏侧性和多发性;按发病年龄可分为儿童型和成人型。
【临床表现】
(一)扭转痉挛(torsiorl spasm)
是全身性扭转性肌张力障碍(torsiorl dystc)nia),临床以四肢、躯干或全身剧烈不随意扭转动作和姿势异常为特征。
可分为:
1.特发性扭转性肌张力障碍儿童期起病的肌张力障碍通常有家族史,出生及发育史正常,多为特发性。
症状常自一侧或双侧下肢开始,逐渐进展至广泛不自主扭转运动和姿势异常,导致严重功能障碍。
.
2.继发性扭转性肌张力障碍成年期起病的肌张力障碍多为散发,可查到病因。
症状常自上肢或躯干开始,约20%的患者最终发展为全身性肌张力障碍,一般不发生严重致残。
体检可见异常运动、姿势,如手臂过度前旋姿势伴屈腕及手指伸展,腿伸直和足跖屈内翻,躯干过屈或过伸等,以躯干为
轴扭转最具特征性;可出现扮鬼脸、痉挛性斜颈、睑痉挛、口一下颌肌张力障碍等,缺乏其他神经系统体征。
(二)局限性肌张力障碍
可为特发性扭转性肌张力障碍的某些特点孤立出现,如痉挛性斜颈、睑痉挛、口一下颌肌张力障碍、痉挛性发音困难(声带)和书写痉挛(一侧上肢)等。
有家族史的患者可作为特发性扭转性肌张力障碍顿挫型,元家族史者可代表成年发病型的局部表现,但成人发病的局限性肌张力障碍也可有家族基础,为常染色体显性遗传,与18p31(DYT7)基因突变有关。
1.痉挛性斜颈(spasmodic torticollis) 是胸锁乳突肌等颈部肌群阵发性不自主收缩引起颈部向一侧扭转,通常30~岁起病,女性较多。
早期常为发作性,后期颈部持续偏向40.一侧。
受累肌肉常有痛感,亦可见肌肉肥大,可因情绪激动而加重,头部得到支持时可减轻,睡眠时消失。
2.Meigc:综合征主要累及眼肌和口、下颌肌肉,表现为睑痉挛和口一下颌肌张力障碍,二者都可作为孤立的局限性肌张力障碍出现,为Meige综合征不完全型,若二者合并出现为完全型。
睑痉挛(blepllarospasril)表现为不自主眼睑闭合,痉挛持续数秒至数分钟。
多为双眼,少数由单眼起病澌波及双眼,精神紧张、阅读、注视时加重,在讲话、唱歌、张口、咀嚼和笑时减轻,睡眠时消失。
口一下颌肌张力障碍(oromandibular dystorlia)表现为不自主张口闭口、撅嘴
和缩拢口唇、伸舌扭舌等。
严重者可使下颌脱臼、牙齿磨损以至脱落,撕裂牙龈,咬掉舌和下唇,影响发声和吞咽等,讲话、咀嚼可触发痉挛,触摸下颌或压迫下颏部可减轻,睡眠时消失。
3.书写痉挛(writer's cramp) 执笔书写时手和前臂出现的肌张力障碍姿势,表现为握笔如握匕首,手臂僵硬,手腕屈曲,肘部不自主地向外弓形抬起,手掌面向侧面等,但做其他动作正常。
本病尚可表现为其他职业痉挛如弹钢琴、打字,以及使用螺丝刀或餐刀等。
药物治疗通常无效,让患者学会用另一只手完成这些任务是必要的。
4.多巴反应性肌张力障碍(Dopa—respc)nsive dystonia,DRD) 或称Seg—aWa病,是常染色体显性遗传疾病,多于儿童期发病(1~9岁),常见于女孩,表现有肌张力障碍性运动和姿势异常,肌张力障碍有明显的日间周期性变化,在下午、傍晚和夜间加重。
对小剂量左旋多巴疗效明显。
患儿也可合并震颤等帕金森综合征的症状和体征。
【诊断要点】
(一)诊断
根据病史、特征性不自主运动和异常姿势通常不难诊断,诊断特发性扭转性肌张力障碍必须排除其他原因继发的肌张
力障碍。
特发性者的实验室检查为正常,而继发性者的实验室检查可发现异常。
鉴别诊断)二(
本病需与面肌痉挛、颈部骨骼肌先天性异常导致的先天性斜颈(K1ippel—Feil畸形)、僵人综合征等鉴别。
【治疗方案及原则】
对继发性肌张力障碍患者首先要进行病因治疗。
对症治疗包括药物、局部注射A型肉毒素、外科治疗。
对局限性或节段性肌张力障碍可首选口服药物或局部注射A型肉毒素,对全身性肌张力障碍宜采用口服药物加选择性局部注射A型肉毒素。
药物或A型肉毒素无效的严重病例可考虑外科治疗。
(一)药物治疗
对扭转性肌张力障碍和局限性肌张力障碍予以药物治疗,可部分改善异常运动。
①抗胆碱能药:盐酸苯海索(安坦)1~2mg,每日3次,逐渐增至可耐受的剂量而达到控制症状。
②地西泮2.5~5mg或硝西泮5~7.5mg,每日3次。
③氟哌啶醇首服每日1次05mg,后逐渐加量至l~2mg,每日3次;氯丙嗪12.5~50mg,每日3次;泰必利100~150mg,每日3 7欠;或舒必利0.1~0.2g,每日3次。
④左旋多多巴反应性肌(巴:用小剂量对一种特发性扭转痉挛变异型张力障碍)有戏剧性效果。
⑤力奥来素(Baclofen)和卡马西平也可能有效。
(二)A型肉毒素
局部注射A型肉毒素(botulinum toxin A)疗效较佳,注射
部位选择痉挛最严重的肌肉或肌电图显示明显异常放电的
肌群,如痉挛性斜颈可选择胸锁乳突肌、颈夹肌、斜方肌等3对肌肉中的4块作多点注射;睑痉挛和口一下颌肌张力障碍分别选择眼裂周围皮下和口轮匝肌多点注射;书写痉挛注射受累肌肉有时会有帮助。
剂量应个体化,疗效可维持3~6个月,重复注射有效。
(三)外科治疗
对严重痉挛性斜颈患者可行副神经和上颈段神经根切断术,部分病例症状可缓解,但可复发。
丘脑损毁术或脑深部电刺激术对某些偏侧肢体肌张力障碍可能有效。