正交优化头孢克肟侧链活性酯的合成工艺
- 格式:docx
- 大小:36.78 KB
- 文档页数:2

头孢克肟活性酯的合成李维;鞠楠【摘要】本文对头孢克肟活性酯的合成进行研究。
最佳工艺为实验温度为-4℃;反应时间为4.5小时;乙腈使用量为150ml;干燥时间为13小时。
【期刊名称】《黑龙江科技信息》【年(卷),期】2012(000)006【总页数】1页(P1-1)【关键词】活性酯;合成;研究【作者】李维;鞠楠【作者单位】哈药集团制药总厂,黑龙江哈尔滨150000;哈药集团制药总厂,黑龙江哈尔滨150000【正文语种】中文【中图分类】TQ465.1头孢克肟是第三代口服头孢菌素,为白色至淡黄色结晶性粉末,易溶于甲醇,几不溶于水。
头孢克肟对化脓性链球菌、肺炎球菌、摩拉卡他菌、肺炎杆菌等具有良好抗菌活性,但对李斯忒菌、多数葡萄球菌、肠杆菌属等无作用。
头孢克肟适用于猩红热、中耳炎、鼻窦炎、肾盂肾炎、尿道炎、胆囊炎、胆管炎等疾病。
本文对头孢克肟活性酯的合成进行研究。
1 实验仪器和试剂1.1 实验仪器SHW110J实验室搅拌机(上海盛海威电气仪表有限公司)、真空泵、布氏漏斗、过滤瓶、2XZ型旋片式真空泵(沈阳万通源泵业制造有限责任公司)、DZF-6210210升立式真空干燥箱(上海和呈仪器制造有限公司)、BILON-5中型冷冻干燥机(北京比朗实验设备有限公司)、WRS-1B数字熔点仪(东莞市全科化玻仪器有限公司)、电子分析天平(德国塞多利斯);PHS一3TC(0.01级)精密数显酸度计(上海天达仪器有限公司)。
1.2 试剂头孢克肟侧链酸(上海泛柯自动化设备有限公司)、二硫化二苯骈噻唑(濮阳蔚林化工股份有限公司)、二氯甲烷(天津市精强化工有限公司)、三乙胺(天津市精强化工有限公司)、亚磷酸三乙酯(天津市精强化工有限公司)、乙腈(天津市精强化工有限公司)。
2 实验方法2.1 实验方案头孢克肟传统合成方法有以下两种:一种以脱乙酰头孢菌素C为原料,经过酰化、氯化等反应制得;另一种以7一ACA为原料,与水杨醛缩合后经过酯化、氯化等反应制得。
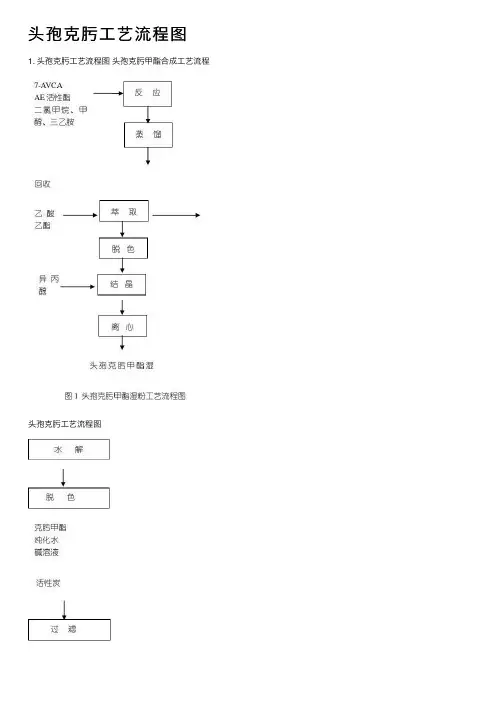
头孢克肟⼯艺流程图
1. 头孢克肟⼯艺流程图头孢克肟甲酯合成⼯艺流程
头孢克肟⼯艺流程图
2. 化学反应过程:头孢克肟酯合成
O
OCH 3
H 2+
N
S
2
N C N
OCH S
N
SH
+
7-AVCA
克肟活性酯
头孢克肟酯
巯基苯并噻唑
头孢克肟酯⽔解
S
N
O
OCH 3
NH 2
O
S
N
N H
O
OH
NH 2
O
O
+
CH 3OH
头孢克肟酯
头孢克肟
甲醇
3. ⽣产原理
将7-A VCA 、⼆氯甲烷、甲醇、三⼄胺、AE 活性酯缩合后,加氯化钠、⼄酸⼄酯静置分相得⽔相,⽔相中加⼊活性炭,加盐酸结晶,得头孢克肟甲酯结晶析出。
在克肟甲酯中加⼊⽚碱溶液,碳酸氢钠溶液⽔解后加⼊活性炭,脱⾊,经三级过滤加盐酸结晶、离⼼、⼲燥、磨粉、分装、包装得成品头孢克肟。
![一种头孢克肟的合成方法[发明专利]](https://uimg.taocdn.com/605e2e500a1c59eef8c75fbfc77da26925c596f6.webp)
[19]中华人民共和国国家知识产权局[12]发明专利申请公布说明书[11]公开号CN 101016305A [43]公开日2007年8月15日[21]申请号200710073334.1[22]申请日2007.02.12[21]申请号200710073334.1[71]申请人河源市制药工程技术研究开发中心地址517447广东省河源市紫金县蓝塘镇立国制药[72]发明人潘行远 [74]专利代理机构深圳市雄杰专利商标代理有限公司代理人王雄杰 王珉[51]Int.CI.C07D 501/22 (2006.01)权利要求书 2 页 说明书 7 页[54]发明名称一种头孢克肟的合成方法[57]摘要本发明公开了一种头孢克肟的合成方法,由于没有用溶媒,省去了对溶媒的回收,节省了费用,降低了生产成本,收率也提高了;另外,本方法没把头孢克肟中间体MECEF结晶出来,而是直接由MECEF的水相萃取液合成头孢克肟,这样既省去了对头孢克肟中间体MECEF的溶解过程,也省去了H 2SO 4和NaHCO 3的使用,还节省了大量的时间,为生产带来了很大的便利,减少了物料的损失,提高了收率;此外,由于把两步反应并为一步反应,实现“一锅煮工艺”,大大地缩短了生产周期,总收率提高到200~210%,含量提高到99%以上。
200710073334.1权 利 要 求 书第1/2页 1.一种头孢克肟的合成方法,其特征在于,步骤如下: (1)在一干净反应罐中加入300~500L氯代甲烷,40~50kgMICA-活性酯,溶解到澄清后加入25~30kgAVNA,纯水100~150L,26~35L 三乙胺,维持温度10~15℃,反应3~6小时;(2)反应毕加入560~680L乙酸乙酯,用3~5℃的纯水300~400L 萃取10~20分钟;(3)静置30~60分钟分液,有机相做好标记,回收装桶;水相用36%的乙酸调PH至6.0~9.0后,加入乙酸乙酯400~450L萃取20~30分钟;(4)静置30~60分钟分液,有机相做好标记,回收装桶;水相加入乙酸乙酯400~450L萃取20~30分钟,静置30~60分钟分液,有机相回收;(5)水相加入乙二胺四乙酸二钠0.5~1kg,连二亚硫酸钠0.5~1 kg,活性炭10~12kg,降温搅拌脱色30~60分钟;(6)压滤,滤饼用水300~340L洗涤,膜过滤,合并滤液和洗液;(7)加入80~100kg碱金属盐溶解,开启夹套冷却水降温至-3~-5℃;(8)快速流加20%NaOH 72~80L,加毕维持-5~-10℃水解10~15分钟,用H P L C检测至显示M E C E F低于0.3%时停止反应;(9)快速流加20%盐酸50~60L,调整PH为5.0~5.5,加入6~8 kg活性炭,搅拌脱色30~60分钟;(10)滤除活性炭,通过微孔滤膜压入另一反应罐中,用冰水洗涤两次,每次100~120L,合并滤液和洗液,开启夹套热水升温,控制温度28~30℃结晶,用20%盐酸调整PH为2.5~2.6;(11)慢速搅拌下降温至0~5℃,维持搅拌1~2小时,放料离心,洗涤,真空干燥得头孢克肟52~63kg。

一种头孢克肟的合成方法头孢克肟(Cefotaxime)是一种第三代头孢菌素类抗生素,具有广谱的抗菌活性,特别适用于治疗多种细菌感染。
本文将介绍一种合成头孢克肟的方法。
头孢克肟的合成主要包括以下几个步骤:底物选择、酰化反应、环化反应、脱保护反应和纯化步骤。
选择适合的底物进行合成。
一种常用的底物是7-氨基硫代醋酸羟乙基酯(7-Aminocephalosporanic acid hydroxyethyl ester,简称7-ACA HE)。
这是一种头孢菌素的前体,能够通过化学反应转化为头孢克肟。
接下来是酰化反应。
将7-ACA HE与氨基甲酸酯(aminoformate ester)反应,生成7-氨基甲酸酯基化合物。
这一步骤通常在碱性条件下进行,以促进反应的进行。
然后是环化反应。
将7-氨基甲酸酯基化合物与环化剂反应,生成头孢克肟的β-内酰胺结构。
这一步骤通常在酸性条件下进行,并通过控制温度和反应时间来提高产率。
脱保护反应是为了去除保护基。
在头孢克肟的合成中,常用的是N-酰基保护基。
通过选择合适的酸性或碱性条件,可以使保护基与头孢克肟分子解离,从而得到纯净的头孢克肟产物。
最后是纯化步骤。
通过结晶、溶剂挥发、析出等方法,可以得到纯度较高的头孢克肟晶体。
晶体经过干燥和粉碎处理后,即可得到头孢克肟的终产物。
在头孢克肟的合成过程中,需要注意控制反应条件和反应时间,以提高产率和纯度。
此外,还需要进行反应中间体的分离和纯化,以确保合成过程的顺利进行。
头孢克肟的合成是一个多步骤的过程,需要合适的底物选择和精确的反应条件控制。
通过合理设计合成路线和反应条件,可以高效地合成出头孢克肟这一重要的抗生素。
头孢克肟的合成方法为医药领域提供了重要的支持,也为广大患者提供了更好的治疗选择。

头孢克肟合成工艺的研究一、本文概述Overview of this article头孢克肟,作为一种重要的β-内酰胺类抗生素,自问世以来就在全球范围内广泛应用于临床治疗各种由革兰氏阳性菌和革兰氏阴性菌引起的感染疾病。
其独特的抗菌机制和良好的治疗效果使得头孢克肟在医药市场上占据重要地位。
然而,随着全球抗生素耐药性的日益严重,头孢克肟的合成工艺研究和优化显得尤为重要。
Cefotaxime, as an important β- Since its inception, lactam antibiotics have been widely used globally in the clinical treatment of various infectious diseases caused by Gram positive and Gram negative bacteria. Its unique antibacterial mechanism and good therapeutic effect make cefixime occupy an important position in the pharmaceutical market. However, with the increasingly severe global antibiotic resistance, research and optimization of the synthesis process of cefuroxime have become particularly important.本文旨在深入探讨头孢克肟的合成工艺,从原料选择、反应条件优化、副产物处理以及工艺经济性等多个方面进行全面分析。
通过对现有合成工艺的综述,提出改进策略和创新思路,以期降低生产成本,提高产品质量,同时满足环境保护和可持续发展的需求。


头孢克肟合成工艺探讨
头孢克肟是一种广谱的第三代头孢菌素类抗生素,广泛应用于治疗呼
吸道、泌尿道、皮肤软组织等感染症。
本文将探讨头孢克肟的合成工艺,
包括原料选择、反应条件、反应步骤等方面。
头孢克肟的合成一般采用核心结构的插入和侧链的引入两步反应。
首先,选择适当的原料进行核心结构的插入反应。
一种常用的方法是采用头
孢菌素C的衍生物作为原料,通过氢氧化钠等碱性条件进行裂环反应得到
α-酮酸中间体,再通过酰化反应引入侧链,形成头孢克肟的前体。
在酰化反应中,选择合适的酸酐和酰化剂是关键。
常用的酸酐有3-
氧代-4-丁酸酐和季戊四酰亚胺,常用的酰化剂有二乙基二硫代氨基甲酸
酯等。
反应条件可根据具体情况进行调整,通常在适宜的温度和pH值下
进行。
头孢克肟的合成过程中还需要特别注意反应条件的控制。
首先,裂环
反应需要严格控制反应温度和时间,以避免产生副产物和降低产率。
其次,在酰化反应中,需要控制酰化剂的用量和反应时间,以提高产物的纯度和
产率。
此外,对于使用的催化剂和溶剂,也需要仔细选择和控制,以确保
反应的顺利进行。
总结起来,头孢克肟的合成工艺包括核心结构的插入和侧链的引入两
步反应。
在具体操作中,需要选择合适的原料、酸酐和酰化剂,严格控制
反应条件,以提高产物的纯度和产率。
此外,还需注意水解反应的条件和
控制,以确保合成过程的顺利进行。
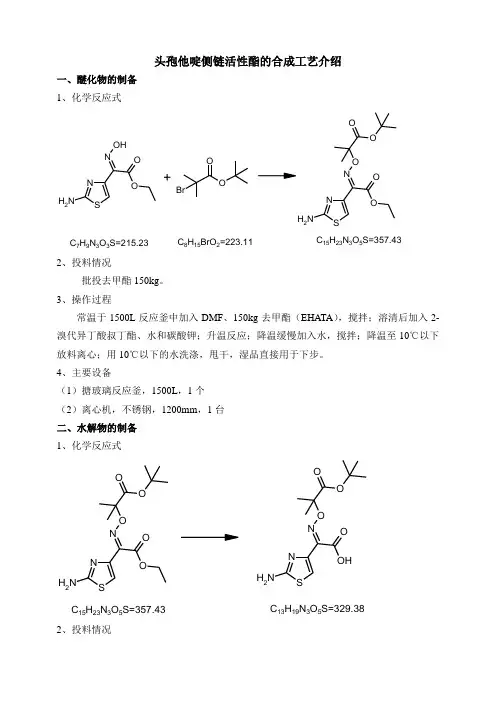
头孢他啶侧链活性酯的合成工艺介绍一、醚化物的制备 1、化学反应式SNNH 2N O OH OSN NH 2N OO OOOOBr2、投料情况批投去甲酯150kg 。
3、操作过程常温于1500L 反应釜中加入DMF 、150kg 去甲酯(EHATA ),搅拌;溶清后加入2-溴代异丁酸叔丁酯、水和碳酸钾;升温反应;降温缓慢加入水,搅拌;降温至10℃以下放料离心;用10℃以下的水洗涤,甩干,湿品直接用于下步。
4、主要设备(1)搪玻璃反应釜,1500L ,1个 (2)离心机,不锈钢,1200mm ,1台 二、水解物的制备 1、化学反应式SNNH 2N OO O OSN NH 2NOHO OO2、投料情况批投上步湿品全量。
3、操作过程常温下于3000L 反应釜中加入水,搅拌下加入氢氧化钠和甲醇;搅拌溶清后加入上步湿品;加入四丁基溴化铵,搅拌约3~4小时;降温滴加浓盐酸,调pH 值;减压回收甲醇;加入保险粉、活性炭和EDTA-2Na ,搅拌;过滤;滤液用浓盐酸继续调pH 值;降温搅拌;离心过滤;滤饼用冰水打浆;离心甩干;滤饼干燥至水份≤7%得到有水酸。
于干燥的1000L 反应釜中加入甲醇和干燥好的有水酸,搅拌;降温,离心过滤,甩干;用甲醇淋洗,甩干;湿品减压干燥至水分小于0.5%以下;出料,称重,得产品约195kg ,收率约82.2%(以去甲酯计)。
4、主要设备(1)不锈钢反应釜,3000L ,一个 (2)离心机,不锈钢,1200mm ,一台 (3)搪玻璃反应釜,1000L ,一个(4)双锥干燥器,不锈钢(或搪玻璃),500L ,一个 三、TAEM 的制备SNNH 2N OHO O OSN NH 2N SON OOSNSNS S2、投料情况批投水解物195kg 3、操作过程于3000L 干燥的反应釜中加入乙腈和苯,搅拌;搅拌下加入195kg 水解物、DM 、吡啶和三乙胺;升温搅拌;滴加亚磷酸三乙酯,搅拌;降温,放料离心,出料;将所得的湿品投入反应锅内,用无水甲醇(或回收甲醇)打浆;放料,用甲醇淋洗,将湿品真空干燥,得产品约240kg ,收率85%左右。

关于头孢克肟中间体合成工艺的改进研究摘要:本文重点就头孢克肟的中间体合成工艺的改进和完善进行了研究,实践发现,此工艺较传统合成工艺不仅收效得到了有效提高,而且大大简化了工艺操作过程。
关键词:头孢克肟中间体合成技术工艺改进随着头孢类药物的不断发展,有关此类药物的研发已经成为我国医药领域的发展重点。
本文先就中间体合成工艺中常用的方法进行了分析,并重点就头孢克肟传统中间体的合成工艺进行了改进和研究。
一、中间体合成工艺中常用方法分析1.溶剂法溶剂法的优点是操作简易、反应剂利用充分、废液产生量低、消耗定额低,且温度稳定。
2.熔融反应熔融反应的优点在于处理过程简便,配比合理,过程产生的三废少,对环境十分有利。
3.催化反应一个良好的催化剂,不仅能加快反应的速度,还能减少生产原材料的浪费,使反应更充分,因此,筛选出合适的催化剂是石油化工生产应当重视的重要环节。
4.电解合成通过电解合成进行生产,可以大大降低生产过程中产生的三废,有效的保护生态环境。
因此,应当充分的利用电解合成。
5.外场反应科技前进的动力很好的推动化学反应技术,运用超声、激光、微波、核辐射以及超高压等高科技可以为化学反应提供力场。
6.生化技术用生物化工的方法可以将乙烯制成环氧乙烷、丙烯制成环氧丙烷、淀粉制成酒精,还可以利用煤制备出甲烷,不对称合成食品添加剂、医药与香料。
此外,还有一些生物制品,例如生物农药、石油蛋白以及生物肥料等。
二、头孢克肟的中间体及其合成工艺作为头孢克肟合成过程中十分重要的侧链,2-(2-氨基-4-噻唑基)-2-(Z)-叔丁氧羰基甲氧亚胺基乙酸活性硫酯的合成对于头孢克肟的生产而言十分关键。
对于次中间体而言,其合成方法很多,多数均是将乙酰乙酸丙烯酯等作为原料,使其同对硝基苄溴发生水解反应而实现的。
对于这类合成工艺来说,有些由于原料较难进行制备,或需要使用价格较为昂贵的钯盐,因此,均无法很好地适合工业的大规模生产。
本文结合多种文献资料,对各种合成工艺的优缺点进行了分析,最终选择了乙酰乙酸甲酯作为合成原料,经过溴化反应得4-溴乙酰乙酸甲酯(1),经亚硝化反应得4-溴-2-羟亚胺基乙酰乙酸甲酯(2),经环合反应得2-(2-氨基-4-噻唑基)-2-(Z)-羟亚胺基乙酸甲酯(3),经醚化反应得2-(2-氨基-4-噻唑基)-2-(Z)-叔丁氧羰基甲氧亚胺基乙酸甲酯(4),经水解反应得2-(2-氨基-4-噻唑基)-2-(Z)-叔丁氧羰基甲氧亚胺基乙酸(5),最后经过硫酯化反应得2-(2-氨基-4-噻唑基)-2-(Z)-叔丁氧羰基甲氧亚胺基乙酸-2-苯并噻唑硫酯(6),这六步实现了头孢克肟中间体合成工艺的改进,实践表明,此合成工艺没有特殊反应,且容易进行操作,所获得的产品质量也相对较好。
![头孢克肟侧链酸活性酯的制备方法[发明专利]](https://uimg.taocdn.com/1018bd2d2cc58bd63086bd5f.webp)
专利名称:头孢克肟侧链酸活性酯的制备方法专利类型:发明专利
发明人:张强,孟令栋,王君伟,刘建国,周忠玉申请号:CN201610747307.7
申请日:20160829
公开号:CN106380467A
公开日:
20170208
专利内容由知识产权出版社提供
摘要:本发明涉及一种头孢克肟侧链酸活性酯的制备方法,属于医药中间体制备技术领域。
所述的头孢克肟侧链酸活性酯的制备方法,是以乙酰乙酸叔丁酯作为起始原料,经肟化、烃化后得到2‑甲氧基甲酰甲氧基亚氨基‑3‑氧代丁酸叔丁酯,采用酰卤作为活化剂,2‑甲氧基甲酰甲氧基亚氨基‑3‑氧代丁酸叔丁酯经成环反应得到中间体(Z)‑2‑(2‑氨基噻唑‑4‑基)‑2‑甲氧基羰基甲氧亚氨基乙酸,然后将该中间体进行酯化反应,得到头孢克肟侧链酸活性酯。
本发明解决了目前工艺中存在的流程长、产品收率低、生产成本高、三废污染高的问题,采用酰基化合物作为活化剂,去掉了收率低、杂质多的卤代过程,缩短了工艺,简化了操作,同时降低了生产成本,减少了三废污染,具有极高的工业应用价值。
申请人:山东金城柯瑞化学有限公司
地址:255074 山东省淄博市高新区四宝山办事处东张村
国籍:CN
代理机构:青岛发思特专利商标代理有限公司
代理人:马俊荣
更多信息请下载全文后查看。

头孢克肟侧链的合成研究的开题报告
头孢克肟是一种广泛用于临床的抗生素药物,对各种细菌感染均有
良好的疗效。
本研究旨在通过合成头孢克肟侧链,寻找更为高效、环保
的制备方法,为头孢克肟的生产提供技术支持。
首先,通过文献调研,深入了解头孢克肟的化学结构及制备方法,
发现头孢克肟的侧链结构较为复杂,制备过程中存在许多难点,如反应
条件的选择、反应物的纯度等问题。
因此,本研究将从反应条件的优化、原料选择等方面展开深入研究。
其次,本研究计划优化头孢克肟侧链的合成方法,通过改变反应条件、使用新型催化剂等手段,提高反应效率和产率,减少副产物的生成。
同时,研究头孢克肟侧链合成过程中的控制方法,保证产品的纯度和质量。
最后,本研究将对头孢克肟侧链在生产中的应用进行验证,考察其
对头孢克肟药物品质的影响。
在此基础上,分析头孢克肟侧链合成方法
的经济效益和环境效益,为头孢克肟的产业化生产提供技术支持。
总之,本研究将通过优化反应条件、改进反应方法等手段,寻找更
为高效、环保的头孢克肟侧链合成方法,为头孢克肟的产业化生产提供
技术支持。
独创性声明本人声明所呈交的学位论文是本人在导师指导下进行的研究工作和取得的研究成果,除了文中特别加以标注和致谢之处外,论文中不包含其他人已经发表或撰写过的研究成果,也不包含为获得天津大学或其他教育机构的学位或证书而使用过的材料。
与我一同工作的同志对本研究所做的任何贡献均已在论文中作了明确的说明并表示了谢意。
学位论文作者签名:签字日期:年月日学位论文版权使用授权书本学位论文作者完全了解天津大学有关保留、使用学位论文的规定。
特授权天津大学可以将学位论文的全部或部分内容编入有关数据库进行检索,并采用影印、缩印或扫描等复制手段保存、汇编以供查阅和借阅。
同意学校向国家有关部门或机构送交论文的复印件和磁盘。
(保密的学位论文在解密后适用本授权说明)学位论文作者签名:导师签名:签字日期:年月日签字日期:年月日摘要头孢他啶是重要的第三代半合成头孢菌素类抗生素品种之一,具有杀菌力强,抗菌谱广的特点,在临床上得到广泛应用,是目前市场上最新的且销售量较大头孢类药物之一。
其上游产品——去甲氨噻肟乙酸乙酯、头孢他啶侧链酸及其活性硫酯限制了头孢他啶药物的发展,由于市场竞争的加剧,生产成本是该药发展的瓶颈。
去甲氨噻肟乙酸乙酯、头孢他啶侧链酸、头孢他啶活性硫酯不仅是合成头孢他啶药物的中间原料,而且也属第四代抗生素中间体,国外己广泛应用,其应用前景将更为广阔。
本论文主要对合成头孢他啶的中间体的生产工艺进行了研究和改进,包括三部分内容:第一部分是关于头孢他啶中间体去甲氨噻肟乙酸乙酯工艺改进。
以乙酰乙酸乙酯为起始原料,在酸性条件下和亚硝酸钠反应生成肟化物,考察了不同配比及肟化反应温度对反应收率和异构反应的影响,再经α-溴代后与硫脲环合生成去甲氨噻肟乙酸乙酯。
通过对比实验确定了经济的合成条件。
并已实现工业化生产,收率达到了59%。
第二部分是关于头孢他啶中间体头孢他啶侧链酸的工艺改进。
去甲氨噻肟乙酸乙酯与α-溴代异丁酸叔丁酯反应,得到醚化产物头孢他啶侧链酸酯,醚化物在碱性条件下选择水解,得到了头孢他啶侧链酸。
一种头孢克肟的合成方法
头孢克肟(Cefotaxime)是一种第三代头孢菌素类抗生素,以
下是一种常见的头孢克肟的合成方法:
步骤1:合成头孢克肟的关键中间体7-氨基-3-(acetoxymethyl)-
3-cephem-4-carboxylic acid(ACTA)。
- 首先,将3-溴代丙酸与丙酰氯在碱性条件下反应,生成3-溴
代丙酰氯(步骤1.1)。
- 然后,3-溴代丙酰氯与7-氨基-3-cephem-4-carboxylic acid在
碱性条件下经Sn2反应结合,生成ACTA(步骤1.2)。
步骤2:将ACTA与三氟乙酸缩合(步骤2.1),然后用三氟
乙酸酐进行酯化反应(步骤2.2),得到7-氨基-3-(acetoxymethyl)-3-cephem-4-carboxylic acid 1,1-dioxide(步骤
2.3)。
步骤3:将7-氨基-3-(acetoxymethyl)-3-cephem-4-carboxylic
acid 1,1-dioxide与丙酰氯在碱性条件下进行酰化反应(步骤
3.1),然后用二乙基氨基氰化亚铜(梯度法)和三甲基乙烯
腈进行底催化反应(步骤3.2),得到头孢克肟(步骤3.3)。
需要注意的是,以上合成方法仅供参考,实际合成过程可能会有所不同。
合成头孢克肟是一项复杂的有机合成工艺,涉及多步反应和中间体的纯化和分离等步骤,需要在严格的实验条件下进行。
在工业生产中,还需要考虑成本、效率和环境等因素。
因此,合成头孢克肟需要专业的有机合成技术和设备支持。
专利名称:一种抗菌素头孢克肟的合成方法专利类型:发明专利
发明人:史利军,孙元强,陈德华
申请号:CN200810020976.X
申请日:20080812
公开号:CN101337969A
公开日:
20090107
专利内容由知识产权出版社提供
摘要:本发明是一种抗菌素头孢克肟的合成方法,以7-氨基-3-乙烯基头孢烷酸(7-AVCA)为起始原料,首先与MICA活性酯作用,分离得到头孢克肟中间体MECEF,中间体MECEF不经分离,直接水解得到头孢克肟产品,将头孢克肟产品合成路线中的缩合和水解合二为一。
优点:具有工艺条件简单、操作方便、产品收率高、产品质量稳定且适合大规模工业化生产,二步反应合二为一,无须分离中间体A,简化操作,缩短了生产周期,收率:200-212%;纯度:99%以上,提高产品的疗效。
将反应中使用昂贵的四氢呋喃改为廉价的丙酮,解决四氢呋喃与其它反应溶剂混合不能回收套用的问题,减少废水的排放,降低生产成本,提高产品的竞争能力。
申请人:苏州万庆药业有限公司
地址:215415 江苏省太仓市富豪经济开发区
国籍:CN
代理机构:南京君陶专利商标代理有限公司
代理人:沈根水
更多信息请下载全文后查看。
正交优化头孢克肟侧链活性酯的合成工艺
梁少娟;刘丹青
【期刊名称】《中国抗生素杂志》
【年(卷),期】2011(036)005
【摘要】By orthogonal test, reaction time, reaction temperature, alkali and the amount of accelerant optimized,the quality of side-chain of cefixime active esterwere improved and the yield was recreased.%通过正交实验,确定反应时间、反应温度、碱量和促进剂的量,并选择适宜条件进行验证实验.改进后提高了头孢克肟侧链活性酯的质量和收率,达到了正交优化实验目的.
【总页数】3页(P351-352,365)
【作者】梁少娟;刘丹青
【作者单位】广州白云山化学制药厂,广州,510515;广州白云山化学制药厂,广州,510515
【正文语种】中文
【中图分类】R978.1+1
【相关文献】
1.正交设计优化光敏剂二氢卟吩e6-C15单甲酯的合成工艺 [J], 单彬;孙渺;孟志;韩贵焱;钱其涵;刘明辉;姚建忠
2.头孢克肟侧链活性酯的合成 [J], 葛洪玉;马卫兴;许兴友
3.头孢克肟侧链(甲酯)活性硫酯的合成 [J], 范美菊;汤沸;杜海生;王勇进
4.头孢克肟侧链活性酯的合成 [J], 朴美兰
5.头孢克肟侧链活性酯的合成 [J], 杜鑫;于洋
因版权原因,仅展示原文概要,查看原文内容请购买。