视神经脊髓炎病因与治疗方法
- 格式:ppt
- 大小:1.42 MB
- 文档页数:15

视神经脊髓炎(neuromyelitis optica,NMO)是由免疫介导的主要累及视神经和脊髓的原发性中枢神经系统炎性脱髓鞘病。
研究发现临床上有一组尚不能满足视神经脊髓炎诊断标准的局限形式脱髓鞘疾病,它们具有与NMO相似的发病机制及临床特征,部分病例最终会演变成NMO。
2007年wingerchuk把上述疾病统一命名为视神经脊髓炎谱系疾病(NMOSD)。
2015年国际NMO诊断小组制定了新的NMOSD诊断标准,取消NMO的单独定义并将其整合入NMOSD的大范畴中。
NMOSD是一组主要由体液免疫参与的抗原-抗体介导的中枢神经系统炎性脱髓鞘疾病谱,其确切病因及发病机制目前仍不明。
大多数患者表现为复发性神经炎和脊髓炎,还可表现为大脑综合征、脑干综合征、最后区综合征等,具有反复发作及致残率高的特点,严重影响患者的生活质量。
该文对近年来NMOSD的研究现状进行梳理报道。
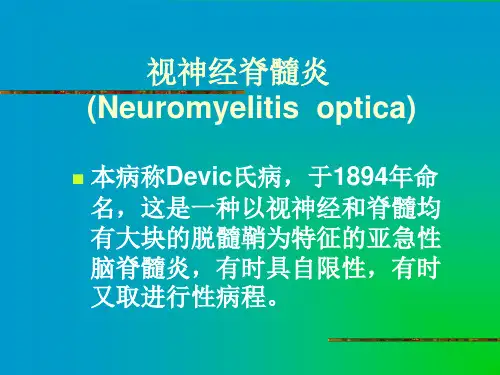
1NMOSD概述1894年Devic和他的博士生Fernand Gault首次描述了视神经脊髓炎(NMO),因此早期称之为Devic病。
当时仍不清楚视神经脊髓炎到底是一种独立的疾病还仅仅是“视神经-脊髓”型多发性硬化(multiple sclero⁃sis,MS)的一种更严重形式。
2004年,之前推测存在的抗原靶位—水通道蛋白4才被证实存在,至此,这两种疾病通过检测AQP4-Abs才被真正区别开。
最新诊断指南将AQP4抗体阳性与抗体阴性两种形式都统一归为NMOSD中。
最近另一种抗原靶位—髓鞘少突胶质细胞已经在NMOSD患者中被检出[1]。
2流行病学和临床特点据报道NMOSD的发病率和患病率与地理位置和DOI:10.16662/ki.1674-0742.2020.30.195视神经脊髓炎谱系疾病刘莎,钱伟东蚌埠医学院第一附属医院神经内科,安徽蚌埠233000[摘要]视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorder,NMOSD)是一种不常见抗体介导的中枢神经系统疾病。

视神经脊髓炎谱系疾病视神经脊髓炎是视神经与脊髓同时或相继受累的脱髓鞘病变。
该病由Devic于1894年首次描述,故又称为Devic 病或Devic综合征。
NMO和多发性硬化均为原发性炎症性脱髓鞘性疾病中的经典疾病,长期以来,NMO与MS的关系存在争议,NMO曾经被划分为MS的亚型。
直至2004年,Lemnon等学者发现了水通道蛋白4抗体后,越来越多证据支持NMO是一种独立于MS的疾病,具有不同的发病机制,需要不同的干预手段以预防复发和延缓神经功能丧失。
但是否存在NMO向MS转化的过渡型仍有待进一步研究。
2015年国际NMO诊断小组(IPND)制定了新的视神经脊髓炎谱系疾病诊断标准,取消了NMO的单独定义,将NMO整合入更广义的NMOSD疾病范畴中。
自此,NMO与NMOSD统一命名为NMOSD。
IPND进一步对NMOSD进行分层诊断,分为AQP4-IgG阳性组和AQP4-IgG阴性组,并分别制定了相应的诊断细则。
病因和发病机制研究究表明,HLA基因仍然是NMOSD的主要易感基因。
然而,东西方患者存在差异,高加索NMOSD可能与HLA-DRB1*03及HLA-DQB1*04基因多态性有关,而亚洲NMOSD与HLA-DPB1*0501等位基因相关。
AQP4抗体(主要为IgGl)在NMOSD发病机制中可能起到重要作用。
AQP4抗体通过血-脑屏障与星形胶质细胞足突上的AQP4结合,激活补体,动员中性粒细胞和嗜酸性粒细胞,造成CNS组织破坏。
然而,目前关于AQP4抗体作用机制有待进一步研究:①20%~30%的NMOSD患者为AQP4-IgG抗体阴性,这部分患者的发病机制如何;②AQP4-IgG如何通过完整的血-脑屏障进入CNS;③AQP4-IgG 是否破坏丰富表达AQP4的外周器官(如肾脏)。
NMOSD病理表现为累及脊髓数个节段的广泛性脱髓鞘与轴索损伤,灰白质均可受累,伴有空洞及坏死。
活动性病变内巨噬细胞浸润,伴随血管周围大量粒细胞、嗜酸性粒细胞,血管周围有明显的免疫球蛋白(主要为IgM)及补体抗原沉积,支持体液免疫在NMOSD发病机制中的作用。
![视神经脊髓炎[德维克]的护理PPT课件](https://uimg.taocdn.com/504f65977e192279168884868762caaedd33baf3.webp)



视神经脊髓炎(1)单时相病程NMO(2)复发型NMO病因及发病机制病因尚不清楚。
临床表现NMO 主要有视神经和脊髓两大组症候,部分患者合并有脑干损害症状。
大约一半的病人以孤立视神经炎起病,其中20%的病人双侧视神经炎;一半的病人以孤立的脊髓炎起病;10%的病人视神经及脊髓同时受累。
视神经症候眼痛、视力下降或失明、视野缺损。
可单眼、双眼间隔或同时发病。
脊髓症候以横惯性脊髓损害较为多见,包括有脊髓相应病变平面以下传导束型深浅感觉、运动障碍及膀胱直肠功能障碍,神经根性疼痛、痛性痉挛,Lhermitte征,高颈段受累者可出现呼吸肌麻痹症候。
脑干症候包括①顽固性呃逆、恶心、呕吐等延髓颈髓交界区受累症状,此表现在NMO 中相对特异,有些病例为唯一首发表现。
②间脑病变可出现嗜睡、困倦、低钠血症等。
辅助检查磁共振成像(MRI)1.头颅MRI:特征性病灶位于下丘脑、丘脑、三脑室、导水管、桥脑被盖及四脑室周围。
延髓病变,常与颈髓病灶相延续。
病变往往不强化。
2.眼部MRI:急性期可见视神经增粗、肿胀,呈长T1、长T2信号,可见“轨道样”强化。
通常双侧视神经均有异常,视交叉及视觉传导通路上可见异常。
3.脊髓MRI:病变常累及3 个或3 个以上椎体节段,为NMO 最具有特异性的影像表现。
NMO 以颈段或颈胸段同时受累最为多见,病变可向上延伸至延髓下部。
病变多位于脊髓中部,累及大部分灰质和部分白质。
急性期多伴有脊髓肿胀并可见强化。
疾病后期部分病例脊髓变细、萎缩、中心空洞形成。
脑脊液检查急性期脑脊液中性粒细胞和嗜酸性粒细胞增多较常见,约13-35%患者细胞数大于50/mm3。
46-75%患者脑脊液蛋白升高。
小于30%的NMO患者脑脊液寡克隆区带可阳性。
血清NMO-IgGNMO-IgG是NMO的免疫标志物,是鉴别NMO与MS的重要参考依据之一,需反复检测。
此外,NMO 患者NMO-IgG 强阳性其复发可能性较大,其滴定度有可能作为复发与治疗疗效的评价指标。

中国视神经脊髓炎谱系疾病诊断与治疗指南一、本文概述视神经脊髓炎谱系疾病(NMOSD)是一种罕见的中枢神经系统自身免疫性疾病,主要影响视神经和脊髓,导致视力下降、肢体无力、感觉异常等症状。
近年来,随着对该疾病的深入研究,越来越多的治疗方法被应用于临床实践。
为了规范我国NMOSD的诊断和治疗,提高患者的生存质量,我们制定了《中国视神经脊髓炎谱系疾病诊断与治疗指南》。
本指南旨在为临床医生提供NMOSD的最新诊断和治疗建议,帮助医生更好地识别和管理NMOSD患者。
本指南基于国内外相关研究和临床实践,结合我国实际情况,对NMOSD的诊断标准、治疗方法、预后评估等方面进行了详细阐述。
本指南也强调了多学科协作在NMOSD诊断和治疗中的重要性,提倡医生与康复科、神经心理科、眼科等多学科团队合作,为患者提供全面的医疗服务。
希望本指南的发布能为我国NMOSD的诊断和治疗提供有益的参考,为患者带来更好的治疗效果和生活质量。
二、疾病定义与分类视神经脊髓炎谱系疾病(NMOSD)是一组以视神经和脊髓为主要受累部位的中枢神经系统自身免疫性疾病。
该疾病谱系涵盖了视神经脊髓炎(NMO)以及其他具有类似临床表现和病理机制的疾病。
NMOSD 的主要特点包括反复发作的视神经和脊髓炎症,以及高血清学阳性率(如抗水通道蛋白4抗体,AQP4-IgG)和高死亡率。
根据临床表现和影像学特征,NMOSD可分为典型NMO和非典型NMOSD。
典型NMO主要累及视神经和脊髓,表现为视力下降、眼痛、肢体无力、感觉异常等。
非典型NMOSD则可能包括其他中枢神经系统的受累,如脑干、间脑、大脑半球等,临床表现多样,可能包括头痛、恶心、呕吐、意识障碍、认知障碍等。
NMOSD的分类主要基于临床表现、影像学特征、血清学标志物以及病理机制。
近年来,随着对NMOSD研究的深入,对疾病的认识和理解也在不断更新和完善,为临床诊断和治疗提供了更加科学的依据。
以上内容仅为概述,具体的疾病定义与分类应根据最新的临床研究和专业指南进行更新和完善。

![视神经脊髓炎[德维克]的护理查房](https://uimg.taocdn.com/605fc738a36925c52cc58bd63186bceb18e8ed74.webp)

视神经脊髓炎的分型全文共四篇示例,供读者参考第一篇示例:视神经脊髓炎(Optic neuritis and transverse myelitis)是一种典型的中枢神经系统炎症性疾病,通常表现为急性或亚急性发作。
视神经脊髓炎是一种独立存在的实体疾病,也可作为自身免疫疾病的表现之一。
根据临床表现和病理生理特点,视神经脊髓炎可以分为多种不同类型。
一、典型视神经脊髓炎典型视神经脊髓炎是指同时发生视神经炎和脊髓炎的病例。
临床上,患者在数天或数周内表现出一侧或双侧进行性视力减退、眼球运动障碍、视野缺损等症状,并伴有脊髓感觉运动障碍。
磁共振成像(MRI)可显示视神经、脊髓、脑部等部位的病变。
典型视神经脊髓炎常见于自身免疫性疾病,如多发性硬化症等。
二、隐匿性视神经脊髓炎隐匿性视神经脊髓炎是指在临床上表现为急性视神经炎或脊髓炎的患者,但在患者病史、体征和实验室检查上无法确诊的情况。
这种类型的视神经脊髓炎常见于局部感染、药物中毒、代谢性疾病等多种疾病引起的急性视神经炎或脊髓炎。
三、亚急性视神经脊髓炎亚急性视神经脊髓炎是指病情呈现亚急性发展,患者症状渐进加重。
临床上,患者可表现为亚急性进行性视力减退、肌无力、感觉障碍等症状。
病理生理特点是脊髓神经元和感觉神经元受损,常导致永久性残留症状。
亚急性视神经脊髓炎常见于感染性疾病、自身免疫性疾病等。
四、复发性视神经脊髓炎复发性视神经脊髓炎是指患者多次发作视神经炎和(或)脊髓炎的疾病。
临床上,患者可能在发作间隔时间较短的情况下出现多次急性或亚急性病情恶化。
复发性视神经脊髓炎具有较高的复发率和发作间隔时间不定性,严重影响患者的生活质量。
该类型的视神经脊髓炎常见于多发性硬化症等自身免疫性疾病。
在临床上,根据病史、临床表现、实验室检查和影像学检查,可以明确不同类型的视神经脊髓炎。
对于不同类型的视神经脊髓炎,治疗方法也有所不同。
及早诊断和治疗对于提高患者的生存率和生活质量至关重要。
随着医学技术的不断进步,对于视神经脊髓炎的诊治越来越准确和有效,希望患者能够早日康复,恢复健康。
2024视神经脊髓炎指南2024年视神经脊髓炎(optic neuritis)指南是基于当前的研究和临床实践,提供最新的指导意见和建议,以便医生和患者能够更好地理解、诊断和治疗这种疾病。
以下是该指南的主要内容。
1.定义和病因:视神经脊髓炎是一种炎症性疾病,主要影响视神经和脊髓。
最常见的病因是自身免疫性疾病,尤其是多发性硬化症(multiple sclerosis,MS)。
其他可能的病因包括感染、实体瘤和其他免疫性疾病。
2.临床表现:视神经脊髓炎的临床表现包括视力减退、眼球疼痛、色觉异常和视野缺损等。
有些病例还可能出现其他神经系统症状,如肢体无力、感觉异常和平衡障碍等。
3.诊断:视神经脊髓炎的诊断主要依靠临床症状、眼底检查、眼压和视野检查等。
必要时,还可以进行脑磁共振成像(magnetic resonance imaging,MRI)、脑脊液检测和视觉诱发电位检查等。
4.鉴别诊断:诊断视神经脊髓炎时,需要排除其他致盲性疾病,如视神经炎、全视盲、视网膜血管阻塞等。
5.治疗:治疗视神经脊髓炎的主要目标是缓解症状、减轻炎症和预防进展。
常见的治疗方法包括激素治疗、免疫抑制剂和病因治疗。
具体的用药方案应根据患者的具体情况和病因确定。
6.预后和复发:大多数患者经过适当治疗后可以恢复视力。
然而,一部分患者可能会在病程中出现复发,需要加强治疗或调整用药方案。
7.其他治疗考虑:除了药物治疗外,视神经脊髓炎患者还可以考虑接受物理治疗、康复训练和心理辅导等,以促进康复和改善生活质量。
总之,2024视神经脊髓炎指南为医生和患者提供了诊断和治疗视神经脊髓炎的最新指导意见和建议。
它有助于促进早期诊断和治疗,提高患者的治疗效果和生活质量。
视神经脊髓炎(neuromyelitis optica , NMO ))是一种免疫介导的 以视神经和脊髓受累为主的中枢神经系统(CNS )炎性脱髓鞘疾病。
NMO ) 的病因主要与水通道蛋白4抗体(AQP4-IgG )相关,是不同于多发性硬 化(MS )的独立疾病实体。
NMO )临床上多以严重的视神经炎(ON )和 纵向延伸的长节段横贯性脊髓炎(LETM )为特征表现,常于青壮年起病, 女性居多,复发率及致残率高。
传统概念的NMO )被认为病变仅局限于视神经和脊髓。
随着深入硏究 发现,NMO )的临床特征更为广泛,包括一些非视神经和脊髓表现。
这些 病更为广泛,包括一些非视神经和脊髓表现。
这些病变多分布于室管膜周 围AQP4高表达区域,如延髓最后区、丘脑、下丘脑、第三和第四脑室周 围、脑室旁、盼月氐体、大脑半球白质等。
AQP4-IgG 的高度特异性进一步 扩展了对NMO )及其相关疾病的硏究。
临床上有一组尚不能满足NMO 诊 断标准的局限形式的脱髓鞘疾病,可伴随或不伴随AQP4-IgG 阳性,例如 单发或复发性ON ( ON/r-ON )、单发或复发性LETM ( LETM/r-LETM \ 伴有风湿免疫疾病或风湿免疫相关自身免疫抗体阳性的ON 或LETM 等, 它们具有与NMO )相似的发病机制及临床特征,部分病例最终演变为 NMO )0 2007年Wingerchuk 等把上述疾病统一命名为视神经脊髓炎谱 系疾病(neuromyelitis opticaspectrum disorders , NMOSD \ 在随后 的观察研究中发现:(1 ) NMO )和NMOSD 在生物学特性上并没有统计 学差异;(2 )部分NMOSD 患者最终转变为NMO) ;《中 视神经脊髓炎谱系疾病诊断与治疗指南》要点( 3 ) AQP4-IgG阴性NMOSD患者还存在一定的异质性,但目前的免疫治疗策略与NMO) 是相似或相同的。
视神经脊髓炎的分型-概述说明以及解释1.引言1.1 概述视神经脊髓炎是一种炎症性疾病,主要影响视神经和脊髓。
它是由于免疫系统的异常反应导致的,可能与感染、自身免疫病等因素有关。
视神经脊髓炎的发病机制尚未完全清楚,但目前认为与免疫系统的异常反应密切相关。
大多数视神经脊髓炎患者表现出针对自身组织的免疫攻击,导致病变和炎症的发生。
这些炎症反应可能会损害视神经和脊髓的神经纤维,造成视力障碍和运动功能障碍。
视神经脊髓炎的临床表现多样化,常见症状包括视力模糊、眼球运动异常、视野缺损、肢体无力、感觉异常等。
这些症状的严重程度和持续时间因个体差异而异,有些患者可能只有轻微的视力障碍,而另一些患者可能会出现严重的残疾。
为了更好地理解和诊断视神经脊髓炎,医学界对其进行了不同的分型。
目前常用的分型方法包括按病程分型、按病变部位分型和按临床症状分型等。
病程分型根据疾病的发展进程将其分为急性、亚急性和慢性三种类型。
病变部位分型根据受累的神经系统部位将其分为视神经型、脊髓型和脑脊髓型等。
临床症状分型则根据患者主要的临床表现将其分为视力障碍型、运动功能障碍型和感觉障碍型等。
诊断视神经脊髓炎需要综合分析患者的临床症状、体征、实验室检查和影像学检查等多种信息。
常用的诊断方法包括脑脊液检查、视力检查、视野检查、MRI等。
脑脊液检查可以检测到炎症反应的指标,而视力检查和视野检查可以评估视神经功能的损害程度。
MRI可以帮助确定病变的位置和范围。
目前针对视神经脊髓炎的治疗方法主要包括免疫抑制剂的应用、激素治疗和对症治疗等。
免疫抑制剂可以抑制异常免疫反应,减少病变的发生和进展。
激素治疗可以减轻炎症反应和控制免疫系统的活动。
对症治疗主要包括症状缓解和功能康复的方法,如视力康复训练、物理治疗等。
综上所述,视神经脊髓炎是一种免疫系统异常反应引起的炎症性疾病,其病程分型、临床表现、诊断和治疗方法各有特点。
深入研究视神经脊髓炎的分型有助于提高对该疾病的认识和诊断水平,并为患者提供更有效的治疗策略。