化妆品中马来酸二乙酯的检测方式
- 格式:docx
- 大小:33.60 KB
- 文档页数:4

《化妆品中硫酸二甲酯和硫酸二乙酯的测定气相色谱-质谱法》(征求意见稿)编制说明广州质量监督检测研究院二〇一六年十一月目录一、工作简况 0二、标准编制原则和主要内容论据 0三、主要试验验证分析,技术经济论证和预期社会经济效益 (8)四、采用国际标准的程度及对比情况 (18)五、与有关的现行法律、法规和强制性国家标准的关系 (19)六、重大分歧意见与处理经过与依据 (19)七、国家标准作为强制性国家标准或推荐性国家标准的建议 (19)八、贯彻国家标准的要求和措施建议 (19)九、废止现行有关标准的建议 (19)十、其他应予说明的事项 (19)一、工作简况(一)任务来源本标准根据国家标准化管理委员会《关于下达2016 年第一批国家标准制修订计划的通知》(国标委综合[2016] 39 号)立项,项目编号为20160667-T-469。
本标准由全国质量监管重点产品检验方法标准化技术委员会归口。
(二)协作单位本标准由广州质量监督检测研究院负责起草。
(三)主要工作过程本标准起草单位在充分收集、认真研究相关标准及资料的基础上,结合实验室的条件和本方法的技术特点,对水基类、乳液类、膏霜类、啫喱类、粉类和蜡基类化妆品产品进行了试验,在论证了方法的灵敏度、准确性、线性范围和应用范围的前提下,通过反复研究和分析,建立了化妆品中硫酸二甲酯和硫酸二乙酯的测定方法;并随机购买了部分市售化妆品进行测定,考察了方法的适用性。
(四)国家标准主要起草人及其所做的工作等本标准起草人:刘冬虹、吴楚森、王斌、王莉、冼燕萍、吴玉銮。
标准起草人负责收集国内外相关检测方法研究并进行对比,开展关键技术研究,撰写标准的主要内容,组织并参与标准初稿的讨论,对相关的技术细节补充完善等。
二、标准编制原则和主要内容论据(一)编制原则本标准是按照GB/T 1.1-2009《标准化工作导则第1部分:标准的结构和编写规则》和GB/T 20001.4-2001《标准编写规则第4部分:化学分析方法》规定的要求进行编写的。

附件10化妆品中乙醇胺等5种有机胺的检测方法1 适用范围本方法规定了测定化妆品中乙醇胺、二乙醇胺、三乙醇胺、二甲胺、二乙胺的离子色谱法。
本方法适用于膏霜、乳、液、粉类化妆品中乙醇胺、二乙醇胺、三乙醇胺、二甲胺、二乙胺的含量测定。
2 方法提要化妆品中乙醇胺等5种有机胺用流动相提取后,经含羧酸功能基的阳离子交换柱分离,电导检测器检测,以保留时间定性,峰面积定量。
对于阳性结果,可用气相色谱-质谱进行进一步确证。
本方法中乙醇胺、二乙醇胺、三乙醇胺、二甲胺、二乙胺的检出限、定量下限及取0.5g样品时的检出浓度和最低定量浓度见表1。
表1 5种有机胺的检出限、定量下限、检出浓度和最低定量浓度物质名称乙醇胺二乙醇胺三乙醇胺二甲胺二乙胺检出限(ng)9定量下限(ng)15 15 30 15 15检出浓度(μg/g)18 18 36 18 18最低定量浓度(μg/g)60 60 120 60 603 试剂和材料除另有规定外,所用试剂均为分析纯,水为一级实验用水。
甲烷磺酸,优级纯。
正已烷。
乙腈,优级纯。
无水乙醇,优级纯。
无水硫酸钠。
乙醇胺,优级纯,纯度≥99%。
二乙醇胺,优级纯,纯度≥99%。
三乙胺,优级纯,纯度≥99%。
二甲胺水溶液,纯度33%。
二乙胺,优级纯,纯度≥99%。
流动相:取甲烷磺酸、50mL乙腈,加水稀释至1L,过滤后备用。
混合标准溶液:分别称取0.1g(精确到0.0001g)乙醇胺、二乙醇胺、二乙胺,及0.2g(精确到0.0001g)三乙醇胺、0.3g(精确到0.0001g)二甲胺水溶液于100mL容量瓶中,用乙腈定容,配成如表2所示浓度的混合标准储备溶液。
吸取储备溶液于100mL容量瓶中,用流动相定容至刻度,摇匀,得到50mg/L乙醇胺、二乙醇胺、二甲胺、二乙胺和100mg/L三乙醇胺混合标准使用溶液,再用流动相稀释混合标准使用溶液配成系列浓度混合标准工作溶液。
表2 5种有机胺混合标准储备溶液及工作溶液浓度物质名称乙醇胺二乙醇胺三乙醇胺二甲胺二乙胺混合标准储备溶液浓度(mg/L)1000 1000 2000 1000 1000混合标准工作溶液浓度(mg/L)12 2 4 2 2 10 10 20 10 10 25 25 50 25 25 50 50 100 50 504 仪器和设备离子色谱仪,具有电导检测器,配色谱工作站。
附件15:化妆品中颜料橙5等5种禁用着色剂的检测方法1 适用范围本方法规定了唇膏、散粉和指甲油类化妆品中酸性黄36(CI 13065)、颜料橙5(CI 12075)、颜料红53:1(CI 15585:1)、苏丹红Ⅱ(CI 12140)和苏丹红Ⅳ(CI 26105)的高效液相色谱测定方法。
本方法适用于唇膏、散粉和指甲油类化妆品中酸性黄36、颜料橙5、颜料红53:1、苏丹红Ⅱ和苏丹红Ⅳ的测定。
2 方法提要样品经溶剂提取后,用高效液相色谱仪分离,二极管阵列检测器检测,以保留时间和紫外-可见光谱图定性,峰面积外标法定量。
必要时,采用液相色谱-质谱联用法进行确证。
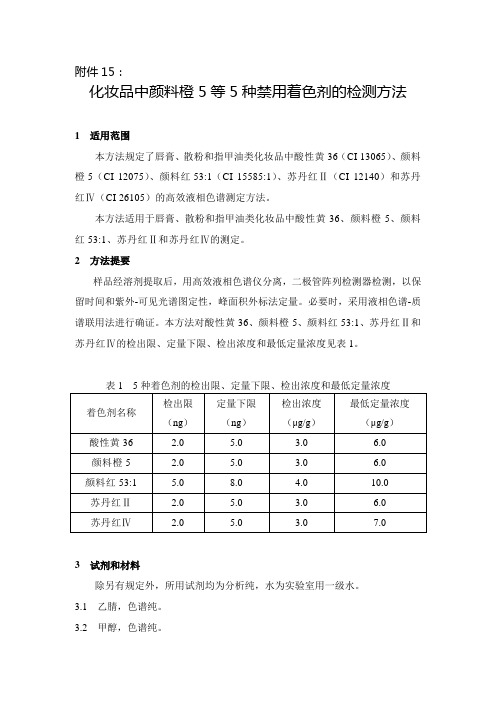
本方法对酸性黄36、颜料橙5、颜料红53:1、苏丹红Ⅱ和苏丹红Ⅳ的检出限、定量下限、检出浓度和最低定量浓度见表1。
表1 5种着色剂的检出限、定量下限、检出浓度和最低定量浓度3 试剂和材料除另有规定外,所用试剂均为分析纯,水为实验室用一级水。
3.1 乙腈,色谱纯。
3.2 甲醇,色谱纯。
3.3 四氢呋喃,色谱纯。
3.4 二甲基亚砜。
3.5 乙醇,色谱纯。
3.6 四丁基氢氧化铵,浓度为55%。
3.7 柠檬酸。
3.8 氨水,浓度为25~28%。
3.9 酸性黄36,纯度≥99%。
3.10 苏丹红Ⅳ,纯度≥94%。
3.11 苏丹红Ⅱ,纯度≥90%。
3.12 颜料橙5,纯度≥97%。
3.13 颜料红53:1,纯度≥95%。
3.14 酸性黄36标准储备溶液(ρ=500 μg/mL):称取酸性黄36(3.9)对照品50 mg(精确至0.1 mg),置于100 mL容量瓶中,用甲醇(3.2)溶解并稀释至刻度,摇匀,即得500 μg/ mL的标准储备溶液。
3.15 苏丹红Ⅳ标准储备溶液(ρ=500 μg/mL):称取苏丹红Ⅳ(3.10)对照品50 mg(按实际含量折算,精确至0.1 mg),置于100 mL容量瓶中,用四氢呋喃(3.3)和乙腈(3.1)混合液(体积比1:9)溶解并稀释至刻度,摇匀,即得500 μg/ mL的标准储备溶液。
![化妆品中邻苯二甲酸酯类化合物的检测方法 国食药监许[2011]96号 附件](https://img.taocdn.com/s1/m/3a4a5efcf705cc175527099b.png)
附件5:化妆品中邻苯二甲酸酯类化合物的检测方法1 范围本方法规定了测定香水、发胶、指甲油等化妆品中10种邻苯二甲酸酯类化合物的高效液相色谱法。
本方法适用于香水、发胶、指甲油等化妆品中10种邻苯二甲酸酯类化合物的含量测定。
本方法所指的10种邻苯二甲酸酯类化合物包括邻苯二甲酸二甲酯(DMP)、邻苯二甲酸二乙酯(DEP)、邻苯二甲酸二正丙酯(DPP)、邻苯二甲酸二正丁酯(DBP)、邻苯二甲酸二正戊酯(DAP)、邻苯二甲酸二正己酯(DHP)、邻苯二甲酸丁基苄酯(BBP)、邻苯二甲酸二环己酯(DCHP)、邻苯二甲酸二正辛酯(DOP)和邻苯二甲酸二异辛酯(DEHP)。
本方法中各种邻苯二甲酸酯类化合物的检出限及取1g样品时的检出浓度见表1。
表1 各种邻苯二甲酸酯类化合物的检出限和检出浓度2 原理邻苯二甲酸酯类化合物在280nm处有特征紫外吸收,可用反相高效液相色谱(HPLC)分离,并根据保留时间和紫外光谱图定性,峰面积定量。
3 试剂3.1 甲醇,色谱纯。
(溶剂应不含邻苯二甲酸酯类化合物)3.2 邻苯二甲酸二甲酯、邻苯二甲酸二乙酯、邻苯二甲酸二正丙酯、邻苯二甲酸丁基苄酯、邻苯二甲酸二正丁酯、邻苯二甲酸二正戊酯、邻苯二甲酸二环己酯、邻苯二甲酸二正己酯、邻苯二甲酸二异辛酯、邻苯二甲酸二正辛酯(纯度>97.5%)。
3.3 混合标准储备溶液(ρ=1 000mg/L):分别称取10种邻苯二甲酸酯类化合物标准品0.05g(精确到0.1mg),用甲醇(3.1)溶解,移入50 mL容量瓶中,定容,摇匀,配成质量浓度为1 000mg/L的混合标准溶液。
4 仪器4.1 高效液相色谱仪,具有二极管阵列检测器,配色谱工作站。
4.2 微量进样器或自动进样装置。
4.3 超声波清洗器。
4.4 高速离心机。
4.5 电子天平。
5 分析步骤5.1 样品预处理称取样品约1g(精确至1mg)于10mL具塞刻度管中,加入甲醇(3.1)至刻度,振摇,超声提取20min,必要时可高速离心。

附件3:化妆品中二乙氨基羟苯甲酰基苯甲酸己酯的检测方法(征求意见稿)1 适用范围本方法规定了采用液相色谱-紫外法测定化妆品中二乙氨基羟苯甲酰基苯甲酸己酯的方法。
本方法适用于膏、霜、乳、液类化妆品中二乙氨基羟苯甲酰基苯甲酸己酯的测定。
本方法二乙氨基羟苯甲酰基苯甲酸己酯的定量下限:0.003μg,取0.1g样品时的定量浓度:300μg/g。
2 方法原理试样采用甲醇超声提取,上清液用液相色谱分离,紫外检测,对化妆品中二乙氨基羟苯甲酰基苯甲酸己酯进行检测,以标准曲线法定量。
3 试剂和材料3.1 甲醇,色谱纯。
3.2 去离子水。
3.3二乙氨基羟苯甲酰基苯甲酸己酯对照品,纯度>99.0%。
3.4二乙氨基羟苯甲酰基苯甲酸己酯标准储备液:取二乙氨基羟苯甲酰基苯甲酸己酯对照品约0.010 g,精密称定,置10 mL容量瓶中,用甲醇(3.1)溶解并定容至刻度,摇匀,配成质量浓度为1.0 g/L的标准储备溶液。
3.5二乙氨基羟苯甲酰基苯甲酸己酯标准工作溶液:精密量取标准储备溶液(3.4)0.1 mL于100 mL容量瓶中,0.1、0.2 mL于20 mL容量瓶中,0.15、0.25、0.4及0.5 mL于5 mL容量瓶中,用甲醇(3.1)稀释至刻度,摇匀。
此时溶液中二乙氨基羟苯甲酰基苯甲酸己酯浓度分别为 1.0、5.0、10.0、30.0、50.0、80.0和100.0 μg/mL。
4 仪器4.1高效液相色谱仪,配紫外检测器。
4.2微型涡旋混合仪。
4.3超声波清洗器。
4.4高速离心机。
4.5分析天平:感量0.0001g。
4.6分析天平:感量0.00001g。
5 测定步骤5.1 样品处理准确称取混匀的试样约0.1g,精确至0.001g,置于50 mL具塞刻度管中,加入20 mL甲醇(3.1),涡旋3 min,振摇,超声提取30 min,必要时可高速离心,精密量取上清液1 mL于10 mL容量瓶中,用甲醇(3.1)稀释至刻度,摇匀,过0.45 μm滤膜,滤液作为待测样液,备用。

化妆品中36种挥发性有机溶剂残留的测定蒋凯;薛晓康;李晓宇【摘要】建立双柱定性-顶空气相色谱质谱联用仪法测定化妆品中36种挥发性有机溶剂残留的方法.使用极性柱VF-1301 ms和非极性柱DB-5 ms两根色谱柱,考察挥发性有机溶剂残留的保留时间以定性,再利用VF-1301 ms柱对存在的残留溶剂进行定量测定.所建立的方法在相应的浓度范围内浓度和峰面积的线性关系良好,线性相关系数为0.999 1~0.999 7,加标回收率在82.0%~117.6%之间,相对标准偏差为1.1%~2.9%,检出限为0.08~29.24 μg/g.该方法检测结果准确、可靠,适用于多种常用化妆品挥发性有机溶剂的同时测定.【期刊名称】《应用化工》【年(卷),期】2019(048)003【总页数】4页(P728-731)【关键词】双柱定性;挥发性有机溶剂;化妆品;顶空气相色谱-质谱法【作者】蒋凯;薛晓康;李晓宇【作者单位】上海化学品公共安全工程技术研究中心上海化工研究院有限公司,上海200062;上海化学品公共安全工程技术研究中心上海化工研究院有限公司,上海200062;上海化学品公共安全工程技术研究中心上海化工研究院有限公司,上海200062【正文语种】中文【中图分类】TQ658;O658随着人们生活水平的提高,化妆品在人类的日常生活中也逐渐占据了不可替代的位置。
而化妆品中普遍存在挥发性有机溶剂残留,这些有机溶剂常用于溶解和分散杀菌防腐剂、香精、表面活性剂油脂及颜料等组分[1-3]。
如长期使用和接触这些有机溶剂,会对人体产生相应的毒害作用,如对皮肤、眼睛和呼吸道造成刺激作用,麻痹和损伤神经,损伤皮脂层等等[4]。
因此,同时对化妆品中多种挥发性有机溶剂残留的测定方法是十分必要的。
然而化妆品成分大多十分复杂,含有多种香精香料,在分析时常常造成干扰。
本文采用双柱定性-顶空气相色谱质谱法[5-15],对36种常见的挥发性有机溶剂进行了测定。
(完整)化妆品原料部分理化指标检测方法编辑整理:尊敬的读者朋友们:这里是精品文档编辑中心,本文档内容是由我和我的同事精心编辑整理后发布的,发布之前我们对文中内容进行仔细校对,但是难免会有疏漏的地方,但是任然希望((完整)化妆品原料部分理化指标检测方法)的内容能够给您的工作和学习带来便利。
同时也真诚的希望收到您的建议和反馈,这将是我们进步的源泉,前进的动力。
本文可编辑可修改,如果觉得对您有帮助请收藏以便随时查阅,最后祝您生活愉快业绩进步,以下为(完整)化妆品原料部分理化指标检测方法的全部内容。
化妆品原料部分理化指标检测方法一、酸值1.定义酸值亦可称为酸价。
中和lg 脂肪酸所需要的氢氧化钾的毫克数,叫做(脂肪酸的)酸值。
油脂的酸值是指中和1g 油脂中的游离脂肪酸所需要的氢氧化钾毫克数。
它们的化学反应为:RCOOH+KOH→RCOOK+H2O已知氢氧化钾的分子量为65。
1,若脂肪酸的分子量为M,中和1g 脂肪酸所需的氢氧化钾毫克数则为:脂肪酸的酸值=65100/M即脂肪酸的酸值与它的分子量成反比.油脂的酸值代表了油脂中游离脂肪酸的含量.油脂存放时间较久后,就会水解产生部分游离脂肪酸,故可用酸值来标志油脂的新鲜程度,酸值愈高,即游离脂肪酸多,表示油脂腐败越利害,越不新鲜,质量越差.一般新鲜的油脂其酸值应在lmg 以下。
2。
测定测定的方法是对于溶解于醇-醚中的规定试样液和对照空白液,用标定过的氢氧化钾液进行滴定至酚酞终点。
(1)试样制备按待测试样酸值的大小(估计),若酸值〈1.0mg,规定取样量5g;若酸值>1。
0mg,规定取样量2g。
将规定试样量放人125ml 锥形烧瓶中,加入25ml 中性乙醇,混合使之溶解,如需要,可在水浴上加热,冷却.另再加入25ml 无水乙醚并混合,如需要可加热,方法如前.即制备好试样液.另配制一个空白试液,不加入试样,只有25ml 无水乙醚和25ml 无水中性乙醇。
(2)滴定试液分别加入lml 酚酞溶液于试样液和空白试液中,再用0。
附件5化妆品中CI11920等13种原料的检验方法CI11920and other12kinds of components1范围本方法规定了高效液相色谱法测定染发类化妆品中CI11920等13种原料的含量。
本方法适用于膏霜乳类化妆品中CI11920等13种原料(不含色淀)含量的测定。
本方法所指的13种原料包括CI11920(食品橙3)、CI12010(溶剂红3)、CI12085(颜料红4)、CI15800(颜料红64)、CI15880(颜料红63)、CI42510(碱性紫14)、CI44045(碱性蓝26)、CI45190(酸性紫9)、CI45370(酸性橙11)、CI47000(溶剂黄33)、CI58000(颜料红83)、CI60725(溶剂紫13)、CI61565(溶剂绿3)。
2方法提要样品提取后,经高效液相色谱仪分离,二极管阵列检测器检测,根据保留时间和紫外光谱定性,峰面积定量,以标准曲线法计算含量。
本方法中各原料的检出限、定量下限及取样量为0.2g,定容至10mL时的检出浓度、最低定量浓度见表1。
表113种原料的检出限、检出浓度、定量下限和最低定量浓度序号着色剂索引号着色剂索引通用中文名检出限(ng)定量下限(ng)检出浓度(μg/g)最低定量浓度(μg/g)1CI11920食品橙30.75 2.57.5252CI12010溶剂红30.75 2.57.5253CI12085颜料红40.30 1.0 3.0104CI15800颜料红640.75 2.57.5255CI15880颜料红630.75 2.57.5256CI42510碱性紫140.30 1.0 3.0107CI44045碱性蓝260.30 1.0 3.0108CI45190酸性紫90.75 2.57.5259CI45370酸性橙110.30 1.0 3.01010CI47000溶剂黄330.30 1.0 3.01011CI58000颜料红830.75 2.57.52512CI60725溶剂紫130.30 1.0 3.01013CI61565溶剂绿30.30 1.0 3.0103试剂和材料除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T6682规定的一级水。
反相高效液相色谱法测定化妆品中的24种防腐剂建立了同时检测化妆品中24种防腐剂含量的反相高效液相色谱法(RP2HPLC)。
采用KromasilC18(4.6mm×250mm,5μm)色谱柱,以磷酸盐缓冲溶液(pH=4.26)为流动相,梯度洗脱。
样品经甲醇超声提取,然后采用RP2HPLC2二极管阵列检测法测定,对样品前处理和色谱条件进行研究和优化。
1引言化妆品中的防腐剂是为了使化妆品在生产、使用和保存过程中免受微生物污染的一类化妆品添加剂。
但大多数防腐剂对人的皮肤会产生不同程度的刺激。
因此,化妆品中防腐剂的用量必须以安全性作为前提。
我国《化妆品卫生规范》对化妆品中防腐剂的使用浓度和范围做了相关的规定。
目前,国内外对化妆品中防腐剂的测定一般多采用高效液相色谱法、气相色谱法、气相色谱质谱法、胶束电动色谱法、毛细管电泳法和伏安法等,而同时测定的防腐剂一般仅为4~8种,最多可同时测定18种,采用的方法均为气相色谱-质谱法。
本实验研究了化妆品中的24种常用防腐剂的样品前处理方法和HPLC分离条件,建立了化妆品中24种常用防腐剂同时检测的HPLC法。
结果表明,本方法简便、快速、准确,应用于实际化妆品中防腐剂的测定,结果满意。
2实验部分2.1仪器与试剂高效液相色谱仪(美国Agilent1100系列),由四元低压泵、柱温箱、二极管阵列检测器及自动进样器组成;KQ-600型超声波清洗仪器(昆山市超声仪器有限公司)。
对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯、水杨酸、5-氯-2-甲基-4-异噻唑啉-3-酮、2-甲基-4-异噻唑啉-3-酮、苯甲醇、苯氧基乙醇、4-氯-3-甲苯酚、三氯生及三氯卡班(Sigma公司);苯甲酸甲酯、苯甲酸乙酯、苯甲酸苯酯及溴硝丙醇(AcrosOrgnics公司);2,4-二氯-3,5-二甲酚、对羟基苯甲酸异丙酯、2-苯酚、4-氯-3,5-二甲酚、对羟基苯甲酸异丁酯及2-苄基-4-氯酚(东京化成工业株式会社);苯甲酸、山梨酸(国家标准物质中心)。
化妆品中地氯雷他定等15种物质的检测方法1 范围本方法规定了采用高效液相色谱-串联质谱法测定化妆品中地氯雷他定(CAS No:100643-71-8)、氯苯那敏(CAS No:132-22-9)、阿司咪唑(CAS No:68844-77-9)、曲吡那敏(CAS No:91-81-6)、溴苯那敏(CAS No:86-22-6)、苯海拉明(CAS No:58-73-1)、异丙嗪(CAS No:60-87-7)、羟嗪(CAS No:68-88-2)、奋乃静(CAS No:58-39-9)、西替利嗪(CAS No:83881-51-0)、氟奋乃静(CAS No:69-23-8)、氯丙嗪(CAS No:50-53-3)、氯雷他定(CAS No:79794-75-5)、特非那定(CAS No:50679-08-8)、赛庚啶(CAS No:129-03-3)等15种抗组胺类物含量的方法。
本方法适用于膏霜、乳液、水剂和啫喱等类型化妆品中地氯雷他定、氯苯那敏、阿司咪唑、曲吡那敏、溴苯那敏、苯海拉明、异丙嗪、羟嗪、奋乃静、西替利嗪、氟奋乃静、氯丙嗪、氯雷他定、特非那定、赛庚啶含量的测定。
2 方法提要以甲醇为溶剂提取化妆品中抗组胺类物质,用高效液相色谱仪分离,质谱检测器检测,采用保留时间和特征离子对丰度比定性,以待测物质相对应离子峰面积定量,以标准曲线法计算含量。
本方法对15种抗组胺类物质的检出限均为1 ng/mL,定量下限均为2 ng/mL,如以取样0.2 g计,检出浓度均为250 ng/g,定量下限浓度均为500 ng/g。
3 试剂和材料除另有规定外,试剂均为分析纯,水为一级实验用水。
3.1 甲醇,色谱纯。
3.2 甲酸,色谱纯。
3.3 甲酸铵,色谱纯。
3.4 甲酸铵-甲酸水溶液:称取甲酸铵(3.3)6.3 g,加1 mL甲酸(3.2),加水1000 mL溶解,用0.22 µm滤膜过滤。
3.5 标准品,参考附录A。
附件1
化妆品中马来酸二乙酯的检测方式
1 适用范围
本方式规定了化妆品中马来酸二乙酯(CAS号:141-05-9)含量的高效液相色谱(HPLC)测定方式和阳性结果确证方式。
本方式适用于乳(霜)、指甲油、香水等化妆品中马来酸二乙酯含量的测定。
2 方式提要
试样在60℃水浴经乙腈超声提取后,过0.45 m滤膜,采纳高效液相色谱系统分离,紫外检测器或二极管阵列检测器进行检测,之外标法定量。
本方式的检出限为μg,定量下限为μg。
若取5.0 g样品,本方式对马来酸二乙酯的检出浓度为mg/kg,定量浓度为mg/kg。
3试剂和材料
除还有规定外,所用试剂均为色谱纯,水为一级水。
乙腈。
马来酸二乙酯标准物质:纯度≥95%。
马来酸二乙酯标准储蓄液(mg/mL):准确称取马来酸二乙酯标准物质0.1 g(精准至mg)于100 mL容量瓶中,乙腈()稀释定容。
马来酸二乙酯系列标准溶液:别离移取标准储蓄液()、、、、、mL于6个100 mL容量瓶中,乙腈()稀释定容,配制成浓度为、、、、、mg/L的系列标准溶液。
4仪器与设备
高效液相色谱仪:配紫外检测器或二极管阵列检测器。
液相色谱-三重四极杆串联质谱仪:配电喷雾离子源。
分析天平:感量别离为mg和mg。
超声清洗器。
涡旋混匀器。
有机微孔滤膜:μm。
具塞比色管,25 mL。
5分析步骤
样品预处置
称取试样5.0 g(精准至1 mg)于25 mL比色管中,加入mL乙腈(),在涡旋混匀器上高速振荡5min。
然
后在60℃水浴中超声提取30min ,静置至20℃(±5℃),用乙腈()定容至刻线,摇匀,过0.45 μm 滤膜,待测。
色谱参考条件
色谱柱:十八烷基硅烷键合硅胶色谱柱,250 mm×4.6 mm (内径),5 μm ;或相当者。
流动相:A 相(乙腈)+B 相(水)=40+60,等度洗脱。
流速: mL/min 。
柱温:30℃。
进样量:10 μL 。
检测波长:220 nm 。
校准曲线的制备
在色谱条件下,别离取系列浓度的标准溶液()进行液相色谱测定,以标准溶液浓度为横坐标,峰面积为纵坐标,绘制校准曲线。
测定
取待测溶液10 μL 注入高效液相色谱仪,依照峰的保留时刻和紫外吸收光谱图定性,依照峰面积从曲线上查出待测溶液中马来酸二乙酯的质量浓度,从而计算样品中马来酸二乙酯的含量。
6 计算
试样中马来酸二乙酯的含量(mg/kg ),按式(1)计算:
D V
w m
ρ=
式中:w ——试样中马来酸二乙酯的质量分数,mg/kg ; m ——称取试样的质量,g ; ρ ——上机溶液浓度,μg/mL ; V ——试样定容体积,mL ; D ——稀释倍数。
7 回收率和周密度
多家实验室验证的平均回收率为%~%,相对标准误差小于5%(n=6)。
8 质谱确证
如检出阳性样品,需经液相色谱-三重四级杆串联质谱法进行阳性确证。
前处置进程见 色谱参考条件
色谱柱:十八烷基硅烷键合硅胶色谱柱色谱柱(150 mm×2.1 mm , μm),或相当者; 流动相:乙腈+水(V:V=75:25); 流速: mL/min ;
柱温:30℃; 进样量:2 L 。
质谱参考条件
离子源:电喷雾离子源(ESI ); 扫描方式:正离子扫描;
监测方式:多反映监测模式(MRM ); 干燥气:N 2; 离子源温度:120℃; 干燥气温度:350℃; 干燥气流速:12 L/min 。
表1 母离子、特点碎片离子、裂解电压及碰撞能
母离子(m/z )
碎片离子(m/z )
裂解电压(V )
碰撞能(V)
173
127
70
5
99
70
5
表2 马来酸二乙酯的定性离子
定性测
定 在相同的实验条件下,样液中被
测物的
色谱峰保留时刻与标准工作液相同,而且在扣除背景后的样液谱图中,所选择的离子对均显现,各定性离子的相对丰度与标准品离子的相对丰度相较,误差不超过表2规定范围内,则可判定样品中存在对应的被测物。
表3 定性确证时相对离子丰度的最大许诺误差
相对离子丰度 >50% 20%~50% 10%~20% ≤10% 允许的相对偏差 ±20%
±25%
±30%
±50%
检出限
本方式对马来酸二乙酯的检出浓度为 mg/kg 。
9 色谱图
名称
定性离子对
定量离子对
丰度比
马来酸二乙酯
>
>
%
>*
图1 马来酸二乙酯标准溶液的高效液相色谱图
(马来酸二乙酯:min)
图2 马来酸二
乙酯标准溶液的
HPLC-MS/MS色谱
图。