高分子化学第七章活性聚合
- 格式:ppt
- 大小:668.55 KB
- 文档页数:40


高分子化学重点名词解释(2)高分子化学重点名词解释p变动还不大,从而使kp/kt增加了近7-8倍,导致加速显著。
(自己总结,仅供参考)动力学链长ν,在聚合动力学研究中,常将一个活性种从引发开始到链终止所消耗的单体分子数定义为动力学链长ν,无链转移时,相当于每一链自由基所连接的单体单元数。
动力学链长应该考虑自初级自由基链引发开始,包括历次链转移以及最后双基终止所消耗的单体总数。
可控/“活性”聚合,传统自由基因为其慢引发、快增长、速终止的机理特性使得聚合物的微结构、聚合度和多分散性无法控制,如果能够降低自由基的浓度或活性,就可减弱双基终止,成为可控/“活性”聚合。
即令活性增长自由基与某化合物反应,经可逆的链终止或链转移,使之转化为低活性的共价休眠种,并使平衡倾向于休眠种一侧,以降低自由基浓度和链终止速率。
调聚反应,当聚合反应体系满足条件kp<<ktr,ka=kp时,聚合速率不变,但聚合物分子量较小,只能获得低聚体,这种聚合反应称为调聚反应。
第四章自由基共聚合无规共聚物是指大分子中两结构单元M1、M2按概率无规律排布,M1、M2连续的单元数不多(一至十几不等)的共聚物;交替共聚物是指M1、M2两单元严格交替相间的共聚物,是无规共聚物的特例;嵌段共聚物是指由较长的M1段链和另一较长的M2段链构成的大分子,每一链段可长达几百至几千结构单元;接枝共聚物是指主链由M1单元组成,支链则由另一种M2单元组成的共聚物。
竞聚率r,在研究共聚物的组成时,将均聚和共聚增长速率常数之比定义为竞聚率r,以表征两种单体的相对活性。
理想共聚合反应(理想恒比共聚):当一对单体满足r1=r2=1的条件时,就会发生理想共聚合反应。
前末端效应,带有位阻或极性较大基团的烯类单体在进行自由基共聚合时,前末端单元对末端自由基的活性将会产生影响,这种影响称作前末端效应。
第五章聚合方法本体聚合是指不加其他介质,单体在少量的引发剂、光或热等的作用下进行的聚合反应;溶液聚合是将单体和少量引发剂溶于适当的溶剂中,在溶液状态下进行的聚合反应;悬浮聚合是借助搅拌并在分散剂的作用下,将不溶于水的单体以小液滴状悬浮在水中进行的聚合反应;乳液聚合是指单体在乳化剂和机械搅拌的作用下,在水中分散成乳液状态进行的聚合反应。


第六章 开环聚合开环聚合属于链式聚合,单体为环化合物,包括环醚、环缩醛、内酯、内酰胺、环硅氧烷等。
7.1 总论7.1.1 环单体的聚合活性环单体的聚合活性由热力学因素和动力学因素共同决定。
1. 热力学因素即环单体和相应的线形聚合物的相对稳定性,它与环大小、成环原子和环的取代基相关。
1) 环烷烃的稳定性:环烷烃进行开环聚合的热力学可行性顺序为:三元环、四元环>八元环>五元环,七元环>六元环。
2) 环的取代基:取代基的引入使聚合热增加、熵变增加,总体使开环聚合可能性降低。
3) 单环单体和多环单体:多环单体的环张力会有所增加,使开环聚合可能性增加。
如8-氧杂[4,3,0]环壬烷,反式的可开环聚合。
4) 成环原子:对于内酯而言,六元、七元环内酯可聚合,而五元环内酯则不可;环三硅氧烷的聚合活性高于环四硅氧烷。
2. 动力学因素环烷烃没有易受活性种攻击的键,因此动力学上仅环丙烷衍生物可进行开环聚合,并且仅能得到低聚物。
环醚、内酯、内酰胺等环单体,因有亲核或亲电子部位,易开环聚合。
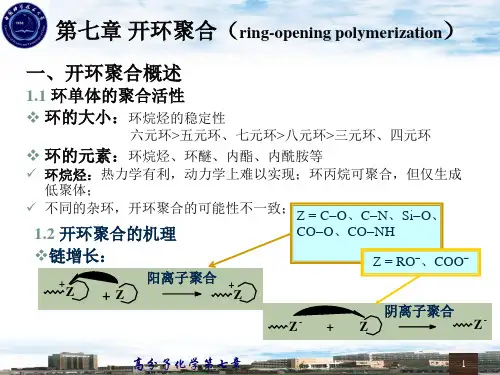
7.1.2 开环聚合机理和特征 1. 聚合机理开环聚合的引发剂为烯烃聚合进行离子型聚合所用的引发剂,引发反应包括初级活性种的形成和单体活性种的形成。
大多数阳离子开环聚合的链增长是通过单体对增长链末端的环状阳离子的亲核反应来进行的,其中的Z 基团为C-O (环醚)、C-N (环氮化合物)、Si-O (环硅氧烷)、酯键和酰胺键;而阴离子聚合的链增长则是增长链末端的阴离子对单体的亲核反应,Z 基团为RO -(环醚)、COO -(内酯)和Si-O -(环硅氧烷)。
;一般情况下,开环聚合的增长链末端带电荷,进行链增长的单体是中性的。
但是,开环聚合还有另一种链增长方式,即所谓的活化单体机理,增长链末端不带电荷,而单体是离子化的,如己内酰胺的阴离子聚合。
2. 开环聚合的基本特征Z-++Z单体加到增长链上进行高分子链的生长;聚合度随转化率增加缓慢,但是在许多场合下呈线性关系;溶剂对聚合反应影响同烯烃的离子聚合;动力学表达式通常类似于链式聚合,特别是活性聚合;许多开环聚合的单体平衡浓度较高,即临界聚合温度较低。