第四章 分子动力学
- 格式:doc
- 大小:34.50 KB
- 文档页数:3


分子动力学
分子动力学(Molecular Dynamics)是运用统计物理学原理,通过计算来研究分子系统中
原子和分子的动态流变,从而对分子间相互作用及对引力法则、量子力学理论和其它物理定律的结果等进行模拟研究的仿真技术。
其基本思想是以细胞原理和迈克尔逊-普朗克动能作为模型基础,借助计算机,通过量子
化学方法理论研究分子在长时间运动中的结构性质及相互作用的力学行为,为原子间的交互作用和分子的动力学运动模拟,可以准确地描述原子性质和反应机理。
在复杂分子系统中,我们可以根据原子间相互作用潜力及其体积影响得出原子间劲度系数。
通过计算,实现分子动力学模拟。
一旦分子动力学模拟被成功应用于实际的物理或有机化学问题,就可以对模拟结果与实验结果进行比较。
将模拟结果与实验结果进行相比较与分析,我们可以更加深入地理解分子的性质。
此外,分子动力学技术还可以用在农业、医学、催化以及合成化学等领域之间。
例如,可以利用此技术来设计新型药物,通过调节抗病毒性和毒性等来减少药物副作用,可以研究加工作用,改进催化剂的性能,优化合成步骤,揭示有机体的生理活动等的究理。
总的来说,分子动力学是一个快速发展的模拟技术,可以模拟和解释小分子和蛋白质等大分子的结构和动态特性,以及丰富科学领域的多种新应用,可以说是一种十分重要的模型。


化学物理学中的分子动力学化学物理学是研究物质中有关化学和物理相互作用的分支学科。
分子动力学则是化学物理学中非常重要的一个方向,它是指利用物理学和数学模型来描述和计算分子的运动行为。
分子动力学能够通过计算机模拟的手段来研究分子在不同温度、压力和环境下的动力学行为及其相互作用。
它是一种基于牛顿力学的数学模拟方法,通常用于研究物质在宏观和微观尺度下的热力学性质和宏观性质。
在分子动力学的研究中,常常使用分子间的势能函数来描述分子间的相互作用和化学反应,基于分子运动规律和动能、势能等物理量对分子进行数值模拟。
这些方法已经得到了广泛的应用,例如在生物化学和纳米技术等领域中,分子动力学已经成为了非常强大的工具。
分子动力学的应用在生物化学领域中,分子动力学可以用于确定生物分子识别和抑制剂的作用机制,如蛋白质、核酸和药物分子等。
分子动力学也可以用来研究分子在溶液中的行为,如蛋白质的折叠和溶剂的影响等。
在材料科学领域中,分子动力学应用非常广泛,如碳纳米管、纳米晶、高分子材料等。
通过模拟不同的反应温度和压力条件下的化学反应,科学家可以预测材料的性能和结构,并为新材料的合成提供理论基础。
另外,分子动力学也在气体动力学中得到了广泛应用,在利用计算机模拟大气层中的气体和气溶胶微粒运动的同时,可以考虑大气环境中的各种复杂作用。
分子动力学的模拟方法晶粒生长晶粒生长是一种分子动力学模拟方法,在晶体过程中使用原子和分子级别的实验数据构建出粒子之间的相互作用,从而通过模拟来预测晶体生长的形貌和性质。
化学反应分子动力学也可以用于模拟化学反应的过程。
这种方法基于分子间的势能,可以模拟分子在反应过程中的能量转移和化学键的形成和断裂。
Nose-Hoover热浴法Nose-Hoover热浴法是一种常用的分子动力学模拟方法,它可以通过在模拟中引入虚拟的热浴,来控制系统的温度和能量波动。
这种方法通常用来模拟大规模分子系统的动力学行为。
总结分子动力学是一种应用广泛的研究方法,它能够模拟分子在不同条件下的运动行为,以及分子间的相互作用和反应过程。



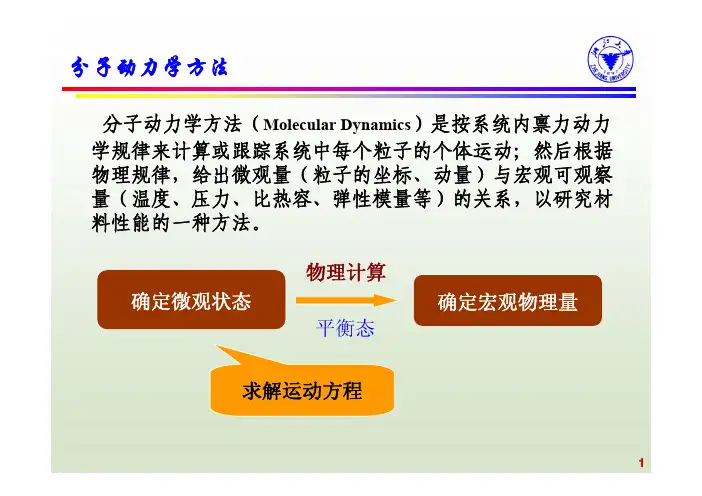
第四章 分子动力学方法§4.1 分子动力学方法第四章 分子动力学方法分子动力学(Molecular Dynamics,简称MD)是模拟大量粒子集合体系(固 体、气体、液体)中单个粒子的运动的一种手法,其关键的概念是运动,即要计 算粒子的位置、速度和取向随时间的演化。
分子动力学中的质点可以是原子、分 子、或更大的粒子集合,只有在研究分子束实验等情况下,粒子才是真正的分子。
与“分子动力学”相类似的名词还有“晶格动力学”(研究固体中原子的振动)和 “分子力学”(分子结构的量子力学),而分子动力学限于模拟经典粒子的运动。
分子动力学简单来说就是用数值方法求解经典力学中的 N 体问题。
自 Newton时代起, N 体问题就被认为是很重要的物理问题,解析求解或质点轨道 的混沌分析是数理力学中的关注点。
但时至今日,该问题重要性的原因已经进化 成,将单粒子动力学与系统的集体状态相联系,人们试图通过考察单个粒子的运 动来解释大量粒子集合系统的行为。
例如,绕过一物体的流体是怎样产生湍流尾 迹的?蛋白质分子中的原子是怎样相互运动从而折叠成生命支撑形态的?流体 气旋怎样产生如木星上的大红斑那样的长寿旋涡的?溶液中的长链分子怎样自 组装成一些特殊结构?等等。
因此,分子动力学在凝聚态物理、材料科学、高分 子化学和分子生物学等许多研究领域都有广泛的应用。
§4.1 分子动力学方法4.1.1 基本概念4.1.1.1 分子动力学分子动力学现已成为分子尺度上模拟的典型方法之一。
它起源于上世纪50 年代,在70年代中开始受到广泛关注。
分子动力学源于自Newton时代以来的古 老概念,即只要知道了系统组分的初始条件和相互作用力,整个系统的行为就可 以计算出来并可以预测。
该自然的决定性力学解释长期左右了科学界。
Laplace 于1814年曾写到:“Given for one instant an intelligence which could comprehend all the forces by which nature is animated and the respective situation of beings who compose it-an intelligence sufficiently vast to submit these data to analysis-it would embrace in the same formula the movements of the greatest bodies of the universe and those of the lightest atoms; for it, nothing would be uncertain and the future, as the past, would be present to its eyes”(现在的 分子动力学模拟中, Laplace的 “intelligence”由计算机实现,“respective situation”即为给定的一组初始条件, “same formula”为算法程序)。



分子动力学langevin摘要:一、分子动力学简介1.分子动力学定义2.分子动力学发展历程二、Langevin 算法在分子动力学中的应用ngevin 算法的原理ngevin 算法在分子动力学模拟中的优势ngevin 算法在分子动力学中的应用案例三、Langevin 算法与其他分子动力学算法的比较ngevin 算法与传统分子动力学算法的比较ngevin 算法与其他随机动力学算法的比较四、Langevin 算法的局限性与未来发展ngevin 算法的局限性ngevin 算法的改进方向正文:一、分子动力学简介分子动力学(Molecular Dynamics,MD)是一种模拟分子运动的数值方法,通过求解量子力学或经典力学方程,对分子之间的相互作用进行模拟。
分子动力学广泛应用于物理、化学、生物等领域,为研究分子结构、分子间相互作用、分子反应等提供了重要手段。
自20 世纪50 年代分子动力学诞生以来,经过数十年的发展,现已成为理论与实验相结合的重要方法。
在计算机技术的推动下,分子动力学模拟已经可以处理较大规模的分子系统,使得许多过去难以研究的科学问题得以迎刃而解。
二、Langevin 算法在分子动力学中的应用ngevin 算法的原理Langevin 算法是一种随机动力学算法,基于Langevin 方程。
在分子动力学中,Langevin 算法通过对系统施加随机力,使得系统在固定的温度和压力下达到平衡。
Langevin 方程描述了粒子在势能曲面上的运动过程,其中包含了弹性碰撞和粘性项。
ngevin 算法在分子动力学模拟中的优势Langevin 算法在分子动力学模拟中有以下优势:(1)可以实现平衡态模拟,适用于多种温度和压力条件;(2)可以处理非弹性碰撞,更接近真实情况;(3)具有较快的收敛速度,可以提高计算效率。
ngevin 算法在分子动力学中的应用案例Langevin 算法在分子动力学中的应用广泛,例如在生物大分子模拟、高分子材料研究、表面物理等领域均有重要应用。
分子动力学分子的运动和相互作用分子动力学是一种研究物质中分子的运动和相互作用的方法。
它通过数值模拟方法,利用经典力学或量子力学的原理模拟分子在时间和空间上的运动,从而揭示物质的宏观性质和微观行为。
本文将介绍分子动力学的基本原理、模拟方法和应用。
一、分子动力学的基本原理分子动力学的基本原理是牛顿第二定律——物体的加速度正比于物体所受的合外力,反比于物体的质量。
对于分子系统来说,可以将每个分子看作质点,其运动由受力决定。
在分子动力学模拟中,通常考虑分子之间的相互作用力,如库仑力、范德华力等,并采用数值积分方法求解运动方程。
二、分子动力学的模拟方法1. 初始构型的设定在进行分子动力学模拟前,需要设定初始构型,即确定分子的位置和速度。
可以根据实验数据或计算结果来设定初始构型,也可以通过随机数生成方法来生成。
2. 动力学方程的数值积分分子动力学模拟需要求解动力学方程,可以采用不同的数值积分方法。
其中,最常用的是Verlet算法和Leapfrog算法。
这些算法通过将时间进行离散化,将运动方程转化为差分方程,并利用迭代方法求解。
3. 相互作用势函数的计算在分子动力学模拟中,相互作用势函数起着至关重要的作用。
常用的相互作用势函数有Lennard-Jones势函数、库仑势函数等。
通过计算相互作用势能,可以获得分子之间的相互作用力,从而模拟分子的运动。
4. 边界条件的设定在分子动力学模拟中,通常需要设定边界条件,以模拟有限的体系。
常用的边界条件有周期性边界条件和固壁边界条件。
周期性边界条件可以模拟无限大的体系,而固壁边界条件则模拟有界的体系。
三、分子动力学的应用1. 材料科学分子动力学可以模拟材料的结构和性质,为材料的设计和开发提供指导。
例如,可以通过模拟纳米材料的热力学性质和力学性能,设计新型材料用于能量存储、传感器等领域。
2. 生物医药分子动力学可以模拟蛋白质、药物和生物大分子的结构和功能,为药物研发和疾病治疗提供指导。
物理化学中的分子动力学在物理化学领域中,分子动力学是一种重要的研究方法,用于揭示分子之间的相互作用和运动规律。
通过模拟和计算分子的运动轨迹,我们可以深入了解物质的性质和行为,为材料科学、生物化学等领域的研究提供有力支持。
一、分子动力学的基本原理分子动力学是基于牛顿力学的一种计算方法,通过求解分子的运动方程,模拟分子在给定条件下的运动轨迹。
其基本原理可以概括为以下几点:1. 分子的力场:分子之间的相互作用力可以通过势能函数来描述,例如分子间的库仑相互作用、范德华力等。
这些力场可以通过实验数据或理论计算得到。
2. 分子的运动方程:根据牛顿第二定律,分子的运动可以由其受到的力和质量决定。
分子动力学模拟通过求解运动方程,得到分子在不同时间点的位置和速度。
3. 时间步长和积分算法:为了模拟分子的运动,需要将时间离散化,即将连续的时间分割为离散的时间步长。
通常使用的积分算法有欧拉法、Verlet算法等,通过迭代计算得到分子在每个时间步长的位置和速度。
二、分子动力学的应用分子动力学在物理化学领域有广泛的应用,以下是几个典型的例子:1. 材料科学:分子动力学可以用于研究材料的力学性质、热传导性能等。
通过模拟材料中原子的运动,可以预测材料的力学响应和热稳定性,为新材料的设计和优化提供指导。
2. 生物化学:分子动力学可以用于研究生物分子的结构和功能。
通过模拟蛋白质、核酸等生物分子的运动,可以揭示其在生物体内的作用机制,为药物设计和疾病治疗提供理论依据。
3. 化学反应:分子动力学可以用于研究化学反应的动力学过程。
通过模拟反应物的运动和相互作用,可以得到反应速率常数、能垒等关键参数,为理解和控制化学反应提供重要信息。
三、分子动力学的挑战和发展尽管分子动力学在物理化学领域有广泛应用,但仍然面临一些挑战和限制。
其中一些包括:1. 计算资源:分子动力学模拟需要大量的计算资源,特别是对于大规模系统和长时间尺度的模拟。
因此,提高计算效率和开发高性能计算方法是当前的研究方向。
第四章 分子动力学方法§4.1 分子动力学方法第四章 分子动力学方法分子动力学(Molecular Dynamics,简称MD)是模拟大量粒子集合体系(固 体、气体、液体)中单个粒子的运动的一种手法,其关键的概念是运动,即要计 算粒子的位置、速度和取向随时间的演化。
分子动力学中的质点可以是原子、分 子、 或更大的粒子集合, 只有在研究分子束实验等情况下, 粒子才是真正的分子。
与“分子动力学”相类似的名词还有“晶格动力学” (研究固体中原子的振动)和 “分子力学” (分子结构的量子力学) ,而分子动力学限于模拟经典粒子的运动。
分子动力学简单 来说就是用数值方法求解经典力学中的 N 体问题。
自 Newton时代起, N 体问题就被认为是很重要的物理问题,解析求解或质点轨道 的混沌分析是数理力学中的关注点。
但时至今日,该问题重要性的原因已经进化 成, 将单粒子动力学与系统的集体状态相联系,人们试图通过考察单个粒子的运 动来解释大量粒子集合系统的行为。
例如,绕过一物体的流体是怎样产生湍流尾 迹的?蛋白质分子中的原子是怎样相互运动从而折叠成生命支撑形态的?流体 气旋怎样产生如木星上的大红斑那样的长寿旋涡的?溶液中的长链分子怎样自 组装成一些特殊结构?等等。
因此,分子动力学在凝聚态物理、材料科学、高分 子化学和分子生物学等许多研究领域都有广泛的应用。
§4.1 分子动力学方法4.1.1 基本概念4.1.1.1 分子动力学分子动力学现已成为分子尺度上模拟的典型方法之一。
它起源于上世纪50 年代,在70年代中开始受到广泛关注。
分子动力学源于自Newton时代以来的古 老概念, 即只要知道了系统组分的初始条件和相互作用力,整个系统的行为就可 以计算出来并可以预测。
该自然的决定性力学解释长期左右了科学界。
Laplace 于1814年曾写到: “Given for one instant an intelligence which could comprehend all the forces by which nature is animated and the respective situation of beings who compose it-an intelligence sufficiently vast to submit these data to analysis-it would embrace in the same formula the movements of the greatest bodies of the universe and those of the lightest atoms; for it, nothing would be uncertain and the future, as the past, would be present to its eyes” (现在的 分子动力学模拟中, Laplace的 “intelligence”由计算机实现, “respective situation”即为给定的一组初始条件, “same formula”为算法程序) 。
分子动力学与分子力学不同,它求解的是随时间变化的分子的状态、行为和过程。
分子动力学将原子看作为一连串的弹性球,原子在某一时刻由于运动而发生坐标变化。
在运动的任一瞬间,通过计算每个原子上的作用力和加速度,来测定它们的位置和运动速度。
由于一个原子的位置相对于其他原子的位置不断变化着,同时力也在变化,可用适当的力场方法,通过评价体系的能量,计算出任一特定原子的力。
分子动力学模拟可作瞬时的、通常为皮秒级(10-12s)的分析,由此模拟计算而获得以一定位置和速度存在的原子的运动轨迹。
计算中根据分子体系的大小、特点和要求来决定模拟时间的长短。
分子动力学方法是一通用的全局优化低能构象的方法。
用分子动力学模拟可使分子构象跨越较大的能垒,因此可以通过升温搜寻构象空间,势能的波动对应着分子构象的变化,当总能量出现最小值时,在常温下(300K)平衡,即可求得低能构象。
在常温下的分子动力学模拟需要很长的时间来克服能量势垒,因此分子动力学对分子构象空间的取样相当缓慢。
提高分子体系的温度,可加大样本分子构型空间的取样效率。
分子动力学计算中,常使用蒙特卡洛算法和模拟退火算法。
蒙特卡洛算法:是一种统计抽样方法。
其基本思想是在求解的空间中随机采样并计算目标函数,以在足够多的采样点中找到一个较高质量的最优解作为最终解。
在动力学计算全局优化低能构象时,以经验势函数随机抽样,不断抽取体系构象,使其逐渐趋于热力学平衡。
该方法需要大量采样才能得到较精确的结果,因此收敛速度较慢。
模拟退火算法:退火是将金属或其他固体材料加热至熔化后,再非常缓慢地冷却的过程。
缓慢冷却是为了凝固成规则的处于最稳态的坚硬晶体状态。
模拟退火算法用于分子动力学计算时,可有效地求得分子的全局优势构象。
过程为:先使体系升温,在高温下进行分子动力学模拟,使分子体系有足够的能量,克服柔性分子中存在的各种旋转能垒和顺反异构能垒,搜寻全部构象空间,在构象空间中选出一些能量相对极小的构象;然后逐渐降温,再进行分子动力学模拟,此时较高的能垒已无法越过,在极小化后去除能量较高的构象,最后可以得到相应的能量最小的优势构象。
模拟退火法的优点在于它能够翻越通常分子动力学条件下不能翻越的能垒;取舍构象时既考虑能量下降的变化,同时也接受部分能量上升的变化,因而能寻找到能量最低点。
此外该方法不依赖于起始构象,消除了人为直觉的偏差。
量子化学:应用量子力学的原理和方法研究研究分子的微观结构,是研究分子结构和性质的最重要的方法之一。
在20年代,科学家用量子力学方法来处理氢分子,奠定了量子化学的基础。
随着量化计算方法的不断发展,计算量及计算速度不断提高,所计算的体系越来越复杂,现在可以计算有机分子甚至较大分子量的生物分子。
运用量化方法借助于计算机可以计算出分子的各种参数,比如分子结构、电子结构、系统总能量和各个轨道的分子信息。
量化计算方法主要分为从头计算法和半经验量化计算。
从头计算法:根据量子力学的基本原理,利用Planck常量、电子质量和电量这三个基本的物理常数以及元素的原子序数,不借助于任何经验参数,计算体系全部电子的分子积分,求解薛定谔方程。
从头算计算结果精度高,可靠性大,但是计算量极大,消耗机时太多,只适用于中等大小的分子体系,对于一些复杂的体系难以处理。
从头算建立在三个基本近似的基础上,即非相对论近似、Born-Oppenheimer近似(绝热近似、核冻结近似)和轨道近似(单电子近似)。
薛定谔方程引入这三个近似后的表达形式为Hartree-Fock-Roothaan方程,HFR方程是计算量化的基础。
进行从头算需选用原子轨道作为基函数,最常用的基函数是STO和GTO,它们不但简化了量化计算所必需的积分,还简化了量化程序化的过程。
HFR方程的求解采用迭代的方法即自洽场Self-Consistent-Field SCF方法,并把对函数迭代转化为对分子轨道组合系数的迭代,得到分子体系得分子轨道、轨道能和波函数,并进一步由波函数计算得到分子体系的其他性质。
如平衡几何构型、电荷密度分布、偶极矩、内旋转和反转势能垒、力常数、势能面和与电子运动有关的能谱等。
求解HFR方程的困难是计算量极大,计算量随基函数数目的4次方递增。
由于从头算引入了3个基本近似,给计算带来一定的误差。
从计算电子相关能出发,可以采用Mφller-Plesset微扰理论(MP)和组态相互作用(Configuration Interaction CI)的校正方法。
量化中引入密度泛函理论,考虑了电子相关性,计算精度提高,时间减少。
密度泛函理论:
半经验算法:基本原理与从头算相同,只是在求解HFR方程时,忽略一些双电子积分或采用实验值拟合的经验参数计算积分值,在计算时仅计算价电子,而将内层电子加入到有效势能中。
虽然这样的处理在理论上不够严密,但大大提高了计算速度,比从头算快100倍以上,所需的磁盘空间和计算机内存也比较小。
因此半经验算法可以用于计算较大的分子体系,只是计算精度较差。
半经验算法主要分为:1)完全不考虑双电子作用的单电子近似方法,如HMO、EHMO等;2)以零微分重叠(zero differential overlap,ZDO)近似为基础的,忽略积分值很小且运算复杂的双电子积分的近似计算方法,如CNDO、INDO、MINDO、NDDO、MNDO、AM1、PM3等。
HMO方法(Huckel Molecular Orbital Method):休克尔分子轨道方法,由Huckel1931年提出,为最简单的半经验计算,能很好地矗立平面的共轭分子。
EHMO方法(Extended Huckel Molecular Orbital Method):扩展的休克尔分子轨道方法,由Hoffmann在1963年提出,是HMO方法的扩展,可用于非共轭体系,处理中考虑全部价电子和所有原子间的相互作用,但完全忽略电子之间的相互作用,没有双电子积分。
该法能成功处理单电子效应占主导地位的问题。
CNDO方法(Complete Neglect of Differential Overlap Method):全略微分重叠方法:由Pople1965年提出,采用微分重叠近似,即不管原子轨道属于分子轨道中的哪一个原子,全部采用ZDO近似。
INDO方法:Intermediate Neglect of Differential Overlap,简略微分重叠方法,由Pople1976年提出,与CNDO近似,作NDO近似,但不忽略但原子微分重叠。
适用于计算键长、键角和偶极矩,以及计算单电子性质的自由基。
MINDO方法:Modified Intermediate Neglect of Differential Overlap,改良的简略微分重叠方法,由Dewar于1969年提出,改法保留了全部单电子排斥积分,能很好地计算分子基态性质,如生成热、键长、键角、偶极矩和第一电离势等。
NDDO: Neglect of Diatomic Differential Overlap,忽略双原子微分重叠方法,由Dewar提出,只忽略双原子微分重叠,与原始的HFR方程最为接近,从理论上并经实验证明计算误差较小,但需计算很多单电子积分,使应用受到限制。
MNDO: Modified Neglect of Diatomic Differential Overlap,改良的忽略双原子微分重叠方法,由Dewar提出,基于NDDO,对生成热、键长、键角、偶极矩和第
一电离势等的计算误差比MINDO等小,但对立体拥挤的分子、氢键和高价化合物等的计算不理想。
AM1方法:Austin Model 1:由Dewar在1985年提出,是对MNDO方法的改良,使用大量实验数据作参量,对基态分子性质的计算明显改善,能正确预测和计算氢键,活化能计算较准确,但计算含磷键、过氧键等误差较大
PM3方法:Parametric Method Number3,参数方法3,是由Steward于1989年提出的又一MNDO的改良方法。
采用新的参量化方法,可以依据生成热、电离热、偶极矩的实验值和计算值分别得到实验与理论参数函数。
对基态分子的计算比MNDO和AM1有较大改进,计算氢键和过氧键误差较小。
由MINDO、MNDO、AM1、PM3四种半经验计算方法编制成的程序MOPAC和AMPAC在量化半经验计算中运用非常广泛。
其中各方法的选用,可以根据具体计算体系的要求,并综合考虑计算精度、计算时间、计算条件等因素来确定。