中日韩人种基因拷贝数变异图谱出炉
- 格式:doc
- 大小:11.52 KB
- 文档页数:2

人类基因组的多样性和进化分析的研究进展在过去的几十年中,人类基因组研究得到了长足的发展,人们越来越认识到人类基因组的多样性和分化的重要性。
这项研究对于我们理解人类起源、进化以及疾病预防和治疗等领域都有着举足轻重的影响。
一、人类基因组的多样性人类基因组是一个十分庞大的系统,由超过20,000个基因所编写。
然而,与其他物种相比,人类的基因组存在非常强烈的多样性。
这些多样性可以体现在基因序列、基因表达和基因组结构等方面。
众所周知,人类有着多样的人种和文化背景,这些背景及其交织的历史对人类基因组造成了深远的影响。
人类基因组研究的进展展示了这些遗传多样性,不仅揭示了人类的起源和演化,而且还为医学研究和功能分析提供了更好的理解。
人类基因组的DNA序列是多样性的一个重要来源。
目前,我们已经测序了许多不同的人类族群和个体,并且将这些序列与参考基因组进行比对。
这些研究揭示出了人类基因组中的许多变异,如单核苷酸多态性(SNP)、插入/缺失和拷贝数变异(CNV)等等。
这些变异能够影响基因的表达水平和功能,有时也会导致疾病发生。
另外,基因组结构的多样性也是人类基因组多样性的一个重要来源。
在不同的个体中,基因的排列和数量可能有所不同。
此外,一些重要的调节元件,在人类基因组中也存在不同的变异,如启动子和增强子等。
这些变异都能够影响基因的表达和功能,从而在人类种群中产生差异。
二、进化分析的研究进展基因组多样性的研究不仅仅是对现代人类多样性的认识,还可以用来理解人类自身的进化历史。
随着高通量测序技术和计算分析工具的不断发展,我们已经突破了以往的研究限制,获取了更详细和准确的人类进化历史信息。
人类的身体和生理特征已经随着时间的推移进行了变化,这些变化往往反映在人类基因组上。
人们进行的大量分析和研究表明,人类的进化历史非常复杂,需要对人类基因组中不同时间和地理位置的变异进行比较。
例如,最近研究发现,从现代人类祖先大约20万年前的非洲开始,最初的现代人类可能分别由不同的祖先群体独立进化而来。

中国北方绵羊基因组拷贝数变异研究侯成林;王伟;周欢敏;张焱如;曹俊伟【摘要】为寻找绵羊基因组中可能的遗传性状相关标记,本试验采用比较基因组杂交(comparative genomic hybridization,CGH)芯片技术,构建了蒙古羊、哈萨克羊、藏羊的拷贝数变异(copy number variation,CNV)多样性图谱.试验结果显示,共检测出28个CNV区域(CNV region,CNVRs),包括11个扩增型、15个缺失型和2个扩增—缺失型.通过功能注释和代谢通路分析发现,在蒙古羊和藏羊基因组中血红蛋白基因存在拷贝数扩张,可能与两种绵羊长期生活在高原低氧环境中产生的适应性有关.对CNVRs和CNV相关基因进行实时荧光定量PCR检验,83.3%的实时荧光定量PCR结果与芯片检测结果一致.通过对中国北方3种绵羊的基因组CNV的研究,为不同绵羊品种间遗传变异的研究奠定了基础.【期刊名称】《中国畜牧兽医》【年(卷),期】2016(043)003【总页数】7页(P784-790)【关键词】绵羊;拷贝数变异;比较基因组杂交;实时荧光定量PCR【作者】侯成林;王伟;周欢敏;张焱如;曹俊伟【作者单位】内蒙古农业大学生命科学学院,呼和浩特010018;包头轻工职业技术学院,包头014030;内蒙古农业大学生命科学学院,呼和浩特010018;内蒙古农业大学生命科学学院,呼和浩特010018;内蒙古农业大学生命科学学院,呼和浩特010018;内蒙古农业大学生命科学学院,呼和浩特010018【正文语种】中文【中图分类】S813在高等生物中,复杂性状一般情况下是由多个等效基因与环境因素共同作用引起的,具有明显的表型复杂性、遗传异质性等特征,一般称为表观遗传性状。
2004年,多个有关“人类基因组计划”研究机构都发现,正常个体间部分基因的拷贝数存在差异的情况[1-2]。
基因拷贝数变异(copy number variation,CNV)主要指与参考基因组相比,由于大片段的插入、缺失或复制所造成的DNA序列的变异。

首个韩国人基因组图谱绘制成功
佚名
【期刊名称】《科学中国人》
【年(卷),期】2008(000)012
【摘要】据韩国联合通讯报道,韩国研究机构日前宣布.第一个完整的韩国人基因组图谱已经绘制成功。
这是人类染色体碱基序列第四次被完整破译,这项成果是由韩国嘉泉医科大学和韩国生命工程研究院的两个研究团队通过共同研究取得的。
研究的目的是建立韩国人标准染色体数据库。
研究人员期待,在染色体碱基序列图谱基础上进一步分析韩国人特有的遗传特性.有助于探明韩国人种发病率较高的遗传性疾病的病因,找到后基因时代的解决办法。
研究还发现,金圣镇的染色体具有323万个单核苷酸多态性位点.
【总页数】1页(P128)
【正文语种】中文
【中图分类】Q987
【相关文献】
1.西藏成功绘制出全球首个青稞基因组图谱
2.我国成功绘制世界首个野生梨基因组图谱
3.西藏成功绘制出全球首个青稞基因组图谱
4.首个染色体水平大蒜基因组图谱绘制成功
5.韩国科学家绘制出首个韩国人基因组图谱
因版权原因,仅展示原文概要,查看原文内容请购买。
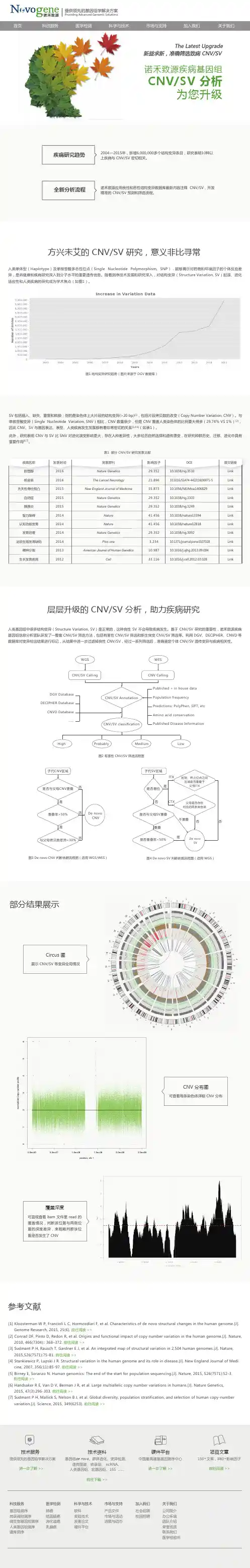
首页 科技服务 医学检测 科学与技术 市场与支持 加入我们 关于我们提供领先的基因组学解决方案Providing Advanced Genomic Solutions参考文献图1 结构变异研究趋势(图片来源于 DGV 数据库)图2 有害性 CNV/SV 筛选流程图图4 De novo SV 判断依据流程图(适用 WGS)图3 De novo CNV 判断依据流程图(适用 WGS/WES)方兴未艾的 CNV/SV 研究,意义非比寻常[1] Kloosterman W P, Francioli L C, Hormozdiari F, et al. Characteristics of de novo structural changes in the human genome.[J]. Genome Research, 2015, 25(6). 前往阅读 >>[2] Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome.[J]. Nature, 2010, 466(7304):: 368–372. 前往阅读 >>[3] Sudmant P H, Rausch T, Gardner E J, et al. An integrated map of structural variation in 2,504 human genomes.[J]. Nature, 2015,526(7571):75-81. 前往阅读 >>[4] Stankiewicz P, Lupski J R. Structural variation in the human genome and its role in disease.[J]. New England Journal of Medi cine, 2007, 356(11):85-97. 前往阅读 >>[5] Birney E, Soranzo N. Human genomics: The end of the start for population sequencing.[J]. Nature, 2015, 526(7571):52-3. 前往阅读 >>[6] Handsaker R E, Van D V, Berman J R, et al. Large multiallelic copy number variations in humans.[J]. Nature Genetics, 2015, 47(3):296-303. 前往阅读 >>[7] Sudmant P H, Mallick S, Nelson B J, et al. Global diversity, population stratification, and selection of human copy-number variation.[J]. Science, 2015, 349(6253). 前往阅读 >>人类单体型(Haplotype)及单核苷酸多态性位点(Single Nucleotide Polymorphism, SNP),能够揭示对药物和环境因子的个体反应差异,是将健康和疾病研究深入到分子水平的重要遗传信息。


中国汉族人群Megsin基因变异与部分多态性位点鉴定【摘要】目的:了解中国汉族人群Megsin基因变异,并对部分多态性位点进行鉴定,筛选适合IgA肾病相关研究的多态性位点. 方法:从基因库中挑选部分Megsin 基因不同功能区域的单核苷酸多态性(SNP)位点,应用聚合酶链式反应限制性片段长度多态性和直接测序的方法,鉴定IgA肾病患者和正常对照组各位点基因型,计算各位点杂合度,根据杂合度大小和疾病组与正常对照组杂合度的差别,筛选适用于IgA肾病相关研究的多态性位点. 结果:在12个从基因库挑选的SNP位点中,6个在我国汉族人群中未发现具有多态性,6个具有多态性. 在第5内含子发现两个新的SNP位点. 在8个确实具有多态性的位点中,3个属少见多态,5个属常见多态,各SNP位点杂合度在IgA肾病组和正常对照组差异无显着性. 结论:中国汉族人群Megsin基因变异与基因库中高加索人群存在较大差异,这可能与中国汉族人群对IgA肾病的高发病率具有重要联系.【关键词】 Megsin;变异;肾小球肾炎,IgA;汉族;中国;多态性,单核苷酸0引言Megsin是一种在肾脏系膜细胞优势表达的基因,在IgA肾病、糖尿病等以系膜细胞增生、系膜基质积聚为主要病理改变的肾脏疾病中,其基因与蛋白表达水平均显着上调[1-2],Megsin转基因小鼠出现与人类IgA肾病相似的肾脏病理改变[3]. 我们以往的研究发现,Megsin基因3′端非翻译区C2093T和C2180T多态性与中国汉族人群IgA肾病的发生显着相关[4].我国汉族人群是IgA肾病的高发人群,其发病率占肾活检人数的30%~40%[5-6],而高加索人群如英国、加拿大和美国白人的发病率明显低于我国汉族人群,在美国仅占肾活检总数的2%~10%[7]. 本研究旨在了解中国汉族人群Megsin基因变异与基因库中高加索人群的差异,并初步筛选适合IgA 肾病相关研究的单核苷酸多态性位点,为最终阐明Megsin基因在IgA肾病发生、发展中的作用提供依据.1对象和方法对象中国汉族IgA肾病患者471例均系我院及香港大学玛丽医院(1997~2004)收治的住院或门诊患者,其中男193例,女278例,均符合WHO关于IgA肾病的诊断标准[8]. 年龄及性别匹配的广东地区既往无肾脏病及其他系统性疾病病史的汉族、健康自愿者200例作为正常对照组.方法收集IgA肾病患者一般临床资料,在患者知情同意的情况下,每名患者及正常对照者抽取外周静脉血2~5 mL, EDTA抗凝. 基因组DNA抽提采用QIAamp DNA kit,严格按照试剂盒说明书操作. 从基因库中调取Megsin基因序列及SNP信息,在Megsin基因7个内含子和唯一一个在基因库中显示有SNP位点的外显子中挑选12个要筛选的位点,随机选取IgA肾病和正常对照标本各50份,应用聚合酶链式反应限制性片段长度多态性和直接测序的方法对各位点的基因型进行鉴定.PCR反应的条件及反应混合物中各成分的浓度,根据位点和引物的不同进行适当调整,各位点所用引物及限制性内切酶. 实验中用于测序的上下游引物分别为5′GGGCCTAGAAAGTGCTGGACACA3′和5′CAAGACACGTTTGGTGGTGTTTCA3′,其PCR 产物长521 bp,测序步骤纯化的DNA模板20 ng, BigDye Terminator Cycle Sequencing Ready Reaction Kitmix(Applied Biosystems, 美国)2 μL,测序引物10 μmol/L组成8 μL反应体系进行PCR测序. 反应条件:98℃ 1 min, 96℃ 30 s, 50℃ 30 s, 60℃ 4 min,25个循环.3 mol/L乙酸钠和无水乙醇沉淀扩增的DNA,再用750 mL/L的乙醇洗涤1次,然后加入18 μL模板变性剂. 测序模板在95℃水浴5 min,立即置于冰水中5 min 将其淬灭成单链,转移至测序管,在ABI3100全自动序列分析仪上进行毛细管电泳测序.表1Megsin基因各SNP位点鉴定所用引物和限制性内切酶统计学处理: 应用软件做统计分析,组间杂合度的比较采用χ2检验,为有统计学意义.2结果IgA肾病患者肾穿刺时年龄(±)岁,病史(±) mo,其他临床指标分别为:收缩压(±) mmHg (1 mmHg= kPa),舒张压(±)mmHg,尿蛋白(±) g/d, 血肌酐(±) μmol/L, 血胆固醇(±) mmol/L,血甘油三酯(±)mmol/L.在从基因库中挑选的12个位点中,9个应用PCRRFLP方法进行基因型鉴定,另3个位点距离较近,用直接测序的方法做基因型鉴定. IgA肾病患者和正常对照者部分多态性位点基因型鉴定的电泳图和测序图. 通。

中国各地人的基因中华文明探源工程初步结果----DNA数据中原官话区西部父系:汉血统90%,阿尔泰P血统5%,棕色人种CD血统5%;中原官话区东部父系:汉血统90%,阿尔泰P血统1%,阿尔泰突变之印第安Q血统1%,棕色人种CD血统5%,越人血统3%;北吴(包括苏南、上海、杭州湾两岸)人父系:汉血统65%,苗瑶族O3d血统10%,棕色人种C血统5%,越人O1血统20%;北吴人母系:越人50%,棕色人种5%,苗瑶族20%,汉人25%。
南吴(不包括苏南、上海、杭州湾两岸)人父系:汉血统40%,苗瑶族(主要是畲族)O3d血统10%,棕色人种C血统10%,越人O1血统40%;南吴人母系:越人60%,棕色人种10%,苗瑶族10%,汉人20%。
广东省广府人父系:越人O1血统40%,秦汉汉族血统10%,宋朝汉族血统50%;广府人母系:越人血统80%,汉族血统20%。
闽南人父系:陈元光系汉族40%,其他汉族10%,越人O1血统40%,棕色人种C血统10%;闽南人母系:越人60%,棕色人种20%,汉人20%。
闽北人父系:王审之系汉族50%,苗瑶族(主要指畲族)O3d血统10%,越人O1血统30%,棕色人种CD血统10%;闽北人母系:越人50%,棕色人种10%,苗瑶族10%,汉人30%。
客家人父系:秦汉汉族10%,宋朝汉族30%,苗瑶族(主要指畲族)O3d血统30%,越人O1血统10%,棕色人种C血统10%;客家人母系:越人30%,棕色人种10%,苗瑶族30%,汉人30%。
湘人(不包括西北部操官话的地区和东部操赣语的地区)父系:赣人(剔除里面的汉血统成分)血统15%,赣人汉血统15%,其他汉血统30%,苗瑶族O3d血统30%,棕色人种C血统10%;湘人(不包括西北部操官话的地区和东部操赣语的地区)母系:越人10%,棕色人种10%,苗瑶族50%,汉人30%。
赣人父系:汉血统50%,苗瑶族O3d血统20%,棕色人种C血统10%,越人O1血统20%;赣人母系:越人30%,棕色人种10%,苗瑶族20%,汉人40%。

临床分子病理实验室二代基因测序检测专家共识近年二代基因测序(next—generation sequencing,NGS)技术快速发展,其应用已进展至临床检测,如遗传疾病、实体肿瘤、血液肿瘤、感染性疾病、人类白细胞抗原分析及非侵袭性产前筛查等。
国内外有关学会已出台相关共识与指南以推动其在临床中的应用。
中华医学会病理学分会和中国抗癌协会肿瘤病理专委会前期组织病理、临床、生物信息等专家进行了充分讨论,拟在NGS的操作流程、数据处理、结果解读等方面作规范和建议,以规范NGS在分子病理领域的应用。
临床分子病理实验室NGS样本可采用甲醛固定石蜡包埋组织(formalin-fixedparaffin—embedded,FFPE)、新鲜组织、各种体液上清液、体液离心细胞块、石蜡包埋标本和血浆/血液标本等。
本共识特色是基于病理评估的组织样本(FFPE、新鲜)的规范。
测序分析范围基于目前临床需求,本共识着重在于目标区域测序(panel)分析的实践。
随着技术的更新和应用的成熟,本共识将持续更新以满足临床需求。
一、实验室总体要求NGS检测实验室的总体设计与要求应参考《分子病理诊断实验室建设指南(试行)》、《医疗机构临床基因扩增检验实验室工作导则》、《个体化医学检测质量保证指南》、《肿瘤个体化治疗检测技术指南》、《个体化医学检测实验室管理办法》、《测序技术的个体化医学检测应用技术指南(试行)》进行。
1。
NGS检测人员的资质要求:NGS检测技术人员应具备临床病理学、分子生物学的相关专业大专以上学历,并经过NGS 技术的理论与技能培训合格。
数据分析人员应具有临床医学或分子生物学或遗传学知识背景并经生物信息学培训。
最终报告应由中级或硕士以上具有病理学背景、经培训合格的本单位执业医师或者授权签字人(医学博士学位或高级职称)审核。
2.NGS检测实验室的区域设置要求:原则上NGS实验室应当有以下分区:样本前处理区、试剂储存和准备区、样本制备区、文库制备区、杂交捕获区/多重PCR区域(第一扩增区)、文库扩增区(第二扩增区)、文库检测与质控区、测序区、数据存贮区.各工作区空气及人员流向需要严格按照《医疗机构临床基因扩增检验实验室工作导则》.分区可根据实际情况合并,但是在前处理和建库时,血液样本应与组织样本分开. 3。

康成生物aCGH芯片技术服务DNA拷贝数变化(CNV)在许多人类疾病(如癌症、遗传性疾病、心血管疾病及糖尿病)的发生发展中起重要作用,但因为CNV 在健康个体间也普遍存在,因此理解特定疾病的分子机制的关键在于鉴定出正常和畸变染色体间的差异。
比较基因组杂交(CGH)是检测基因组DNA的片段扩增或缺失的有效方法,而基于微阵列技术的比较基因组杂交(aCGH)通过在一张芯片上用标记不同荧光素的样品(肿瘤样品和对照样品)同时进行杂交可检测样本基因组和对照基因组间DNA拷贝数变化(CNV),常用于肿瘤及遗传性疾病全基因组CNV检测,直观地表现出肿瘤及遗传性疾病基因组DNA在整个染色体组的缺失或扩增,对肿瘤而言缺失部位可能包含抑癌基因,而扩增片段则可能存在致癌基因。
●什么是拷贝数的变异(CNV)?人类的基因组是由包括60亿个化学碱基(或者称为核苷酸)的DNA组成的,并被包装到23对染色体中,每对染色体都是一条来自父代,一条来自母代。
这些DNA编码大约30,000个基因。
过去通常认为基因在基因组中是以2个拷贝的形式存在,然而目前的发现已经表明:有大量的DNA片段在拷贝数上有很大的变异,这些DNA片段的大小从数千到数百万碱基不等。
这些拷贝数的变异(简称CNVs)包含拷贝数改变的基因,比方说,那些过去认为每对染色体上存在2个拷贝的基因,现在发现有时是1个拷贝,有时是3个甚至3个以上,在少数罕见的情况下这些基因还会一起缺失。
●为什么说CNVs很重要?染色体上DNA序列的不同决定了我们个体的独特性,包括对疾病的易感性等等。
过去认为DNA的单核苷酸的改变(称为SNPs单核苷酸多态性)是基因变异的主要形式,最近的研究发现CNVs的发生率至少是SNPs的3倍。
因为CNVs中经常包含一些在人类疾病发生和对药物的反应中起着重要的作用的基因,所以理解CNV形成机制能够帮助我们更好地理解人类基因组的进化。
●新的CNV图谱有什么帮助?新的CNV图谱能够在三个领域对医学研究有帮助:首先也是最重要的,就是能够帮助寻找与常见疾病相关的基因,到目前为止,还没有真正认识到这些基因的CNVs在人类健康中所起的作用,我们正试图证明这一点;第二,CNV图谱正被用来研究家族遗传病;第三,数千种严重的发育缺陷正是由于染色体的重排引起的,CNV图谱被用来排除在未受影响个体上变异的发生,帮助研究人员确定可能涉及到的目标区域,整个数据的生成要归功于一个更加精确及完整的人类基因组参考序列。

第 卷第 期海洋与湖沼∂年 月 ≤∞ ∞× ≥ ≤ ≥皱纹盘鲍中国群体和日本群体的自交与杂交Φ1的ΡΑΠ∆标记3张国范王继红 赵洪恩 阙华勇刘晓中国科学院海洋研究所青岛大连水产学院大连大连市水产研究所大连提要利用皱纹盘鲍中国群体和日本群体野生个体的单自交和单正反杂交获得了 个ƒ家系 其中家系 ≤! ≤≤!≤≤ !⁄ 分别为日本α≅中国⎯!中国α≅中国⎯!中国α≅日本⎯和日本α≅日本⎯组合 采用 个引物对上述四个家系及其各自亲本个体的遗传结构进行了°⁄分析∀结果表明 四个家系的平均杂合度 ≤为 ≤≤为 ≤≤ 为 ⁄为 各家系两亲本间遗传距离 ≤为 ≤≤为 ≤≤ 为 ⁄ 为 ∀各家系子代群体与父母本的遗传距离 ≤为 和 ≤≤为 和 ≤≤ 为和 ⁄ 为 和 ∀各家系子代个体间的遗传距离 ≤为 ≤≤为≤≤ 为 ⁄ 为 而子代各群体间遗传距离均小于 ∀关键词皱纹盘鲍 不同地理群体 杂交子代 杂交优势 °⁄分子标记中图分类号±皱纹盘鲍 Ηαλιοτισδισχυσηανναι 是我国海珍品养殖的重要种类∀近年来由于频繁发生病害 张国范等 给其养殖产业造成了重大损失∀为了提高养殖鲍抗逆性和加快其生长速度 /杂种优势0的利用在生产实践中有很大的应用价值∀遗传育种的理论与实践都已证明杂交优势利用的关键在于找到强优势的杂交组合 亲本间遗传差异大时 它们的杂交优势亦强∀遗传差异的本质是⁄ 序列水平的差异∀用⁄ 分子生物学方法能正确地测定亲本间的分子差异或分子遗传距离 从而可作为预测杂交种优势的重要指标 帮助育种学家减少配制杂交组合时的盲目性∀分析和比较不同皱纹盘鲍群体间个体及其杂交子代的遗传结构是预测杂种优势的重要途径之一 而 °⁄标记技术为此提供了有效的手段∀°⁄标记现已用于动植物遗传作图!基因快速定位!特殊染色体区段的鉴定和分离!性别鉴定以及群体遗传变异的研究 季维志等 张细权等 梁利群等 滕春波等 宋林生等 孙致良等 石拓等 孙效文等 孙易等 张四明等 李斌等 刘萍等 ∏ √ εταλ εταλ3国家 资助项目 号 国家杰出青年科学基金资助项目 号∀张国范 男 出生于 年 月 博士 研究员 ∞2收稿日期 2 2 收修改稿日期 2 2期张国范等 皱纹盘鲍中国群体和日本群体的自交与杂交ƒ 的 °⁄标记∀近期其在杂种优势预测方面的应用又引起越来越多学者的关注 董在杰等 吕雪梅等 许明辉 李祥龙等 ∀对于海洋贝类 进行⁄ 多态性研究的比较多 主要用于种内与种间不同群体间的变异与分化!种质鉴定与系统进化研究等 刘必谦等 εταλ εταλ εταλ εταλ ∀在预测杂种优势等有关分子标记辅助育种方面的研究还未见报道∀本文中利用 °⁄技术对中国野生与日本野生皱纹盘鲍个体自交及正反杂交获得的 个子一代家系及其各自亲本个体的遗传结构进行了 °⁄研究 目的在于从分子水平上了解这 个家系及其父母亲本个体的遗传结构 为杂交育种的生产实践提供分子遗传学基础 为皱纹盘鲍种质资源的更好利用提供基础资料和科学依据 促进鲍杂交育种中的杂种优势预测及分子标记辅助育种工作的开展∀1材料与方法1 1材料1 1 1实验用鲍实验用皱纹盘鲍 Ηαλιοτισδισχυσηανναι 于 年 月分别采自辽宁大连长海县獐子岛和日本岩手县∀取野生中国和日本皱纹盘鲍雌雄亲本各 只进行杂交及自交 产生 个交配组合及其ƒ 家系 ≤ 日本α≅中国⎯ ≤≤ 中国α≅中国⎯ ≤≤ 中国α≅日本⎯ ⁄ 日本α≅日本⎯∀各家系ƒ 饲养至壳长 左右∀上述工作均在大连市水产研究所的协助下完成∀1 1 2仪器与试剂°≤ 扩增仪为°∞公司生产的°∞2 型∀引物!× ⁄ 聚合酶! ×°等试剂均购自上海≥ 公司∀1 2方法1 2 1基因组⁄ 的提取按萨姆布鲁克等 的方法略做改进∀将各家系的亲本野生鲍及其 个ƒ 个体活体取足部肌肉组织固定于 乙醇中 取 组织用蒸馏水清洗!切成碎沫并溶于 Λ 裂解液中 成分 × ≤ ∞⁄× ≥⁄≥ ∀加入蛋白酶 至终浓度为 Λ 来回颠倒离心管数次 置于 ε水浴中消化 在此期间转动离心管数次 以使之充分消化 ∀将上述消化液冷至室温 加等体积的酚抽提一次 以 转 离心 取上清液 再分别用等体积的酚 氯仿 酚Β氯仿Β异戊醇 Β Β 及氯仿 氯仿Β异戊醇 Β 各抽提一次 离心条件同上 取上清液∀向上清液中加入双倍体积的冷的 无水乙醇和十分之一体积的 ε冷冻 再用 乙醇洗涤⁄ 沉淀 次 沉淀置于室温凉干 ×∞溶解 ε保存∀1 2 2⁄ 定量使用 ∞≤ 公司生产的⁄ 型紫外分光光度计 将提取的模板⁄ Λ 稀释 倍至 Λ 进行测定∀在 时读数用于计算样品中核酸的浓度 ⁄值等于 时 相当于大约 Λ 双链⁄ 同时读取 ⁄ ⁄ 的比值 以估计所提⁄ 的纯度 纯⁄ ⁄ ⁄ 为 ) ∀同时辅以凝胶电泳肉眼观察所提⁄ 的完整性∀将⁄ 原液稀释到 Λ 于 ε保存待用∀1 2 3 °⁄条件 °⁄程序 ε预变性 后 ε变性 ε复性 ε延伸 个循环∀最后 ε延伸 ε保存∀ °⁄反应总体积为 Λ 其中含有 ÷ °⁄反应缓冲液 Λ ≤ ×° 引物 × 酶 基因海洋与湖沼 卷组⁄ ∀1 2 4电泳及成像扩增产物在 的琼脂糖凝胶中电泳 电压为 ∂ 左右∀电泳完成后以 ∂°公司的 ⁄≥ 型凝胶成像仪对凝胶进行观察!拍照及保存 并以 2 ° 软件包对图片进行计带处理 有带记为 无带记为 并对每个扩增带进行分子量的估算∀1 2 5数据处理各群体的平均杂合度∀纯合度 ϑ (ΕΞ ι)/ν,杂合度:Η ϑ∀其中:Ξι表示第ι个等位基因的频率,ν为测定位点数(季维志等, )∀遗传分化指数(ΓΣΤ)∀总群体的基因多样性(ΗΤ)分解为群体内基因多样性(ΗΣ)和群体间基因多样性(∆ΣΤ):ΗΤ ΗΣ ∆ΣΤ,ΓΣΤ ∆ΣΤ/ΗΤ∀这里ΗΣ ϑΣ (Εϑι)/Σ,其中Σ为群体的数目∀ϑι (ΕΞ ικ)/ν,这里ϑι是第ι个群体内的基因一致性,Ξικ是第ι个群体内第κ个等位基因的频率,ν是ι个群体的基因位点数目;∆ΣΤ (ΕΕ∆ιϕ)/Σ ,这里∆ιϕ (ϑι ϑϕ)/ ϑιϕ,其中ϑιϕ是第ι个和第ϕ个群体间的基因一致性:ϑιϕ (ΕΞικΞϕκ)/ν∀群体间的遗传距离∀根据 的计算方法计算∀∆ Ι,这里Ι ϑΞΨ/ (ϑΞϑΨ) / ,其中ϑΞ!ϑΨ和ϑΞΨ分别是所有位点上ϕξ!ϕψ和ϕξψ的算术平均值∀这里ϕξ ΕΞι,ϕψ ΕΨι,ϕξψ ΕΞιΨι,Ξι!Ψι分别是Ξ!Ψ群体中第ι个等位基因的频率∀群体内遗传距离∀随机扩增多态⁄ 片段的共享度根据下列公式 εταλ 计算 Φ {Ε[ Νξψ/(Νξ Νψ)]}/ν,Π Φ∀式中,Φ为相似率,Νξ和Νψ分别为个体ξ和个体ψ的随机扩增多态⁄ 位点数 Νξψ为两个体间相同的位点数,ν为个体间相比较的两两配对数,Π为遗传距离∀多态率∀多态率π ξ/ρ,其中ξ为具有多态性的位点,ρ为扩增出的总位点数∀2结果2.1ΡΑΠ∆扩增结果在 °⁄扩增中 所用引物为经过挑选的扩增谱带清晰稳定的 条引物 ≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ !≥ ∀选择扩增片段大小在 ) 之间的位点进行统计分析∀结果共检测到 个位点 平均每个引物扩增出 个位点∀其中 ≤的多态位点数为 个 多态率为 ≤≤的多态位点数为 个 多态率为 ≤≤ 的多态位点数为 个 多态率为 ⁄ 的多态位点数为 个 多态率为 ∀多态位点的显性频率为 家系 ≤在 ) 之间 家系 ≤≤在 ) 之间 家系≤≤ 在 ) 之间 家系⁄ 在 ) 之间∀各引物的扩增情况见表 ∀扩增结果显示 不同引物得到的扩增片段数各不相同 所揭示的多态性也不尽相同∀图 显示的是引物≥ 对 个家系的扩增结果 可以看出扩增位点数虽然居中 却显示很高的多态性 并且在 ! ! 处呈现子代所扩增的谱带强度比亲本大为增强的现象∀图 是引物≥ 对 个家系的扩增结果 可以看出扩增位点极多 且几乎皆为单态∀另外从整体的 °⁄扩增情况来看 亲本出现的谱带都能在子代中找到 并且有的像父本 有的像母本 有的是两者基因型的综合 符合典型的孟德尔共显性遗传规律∀表1各引物序列及四个家系的ΡΑΠ∆扩增情况× × . ∏ °⁄ ∏引物序列 .) .产物的总片段数产物的多态数≤ ≤≤≤ ≤⁄ ≤ ≤≤≤ ≤⁄≥ × ×≤ ≤ ≥ ×≤× × ≥ ≤≤ ≤×× × ≥ × × ≤≤≤≤ ≥ ≤ ≤ ×≤ ≥ ××≤≤≤≤≤≤ ≥ ≤ ×≤≤ ≤ ≥ × × ≤× ≥ × ≤ ≤ ≥ ≤× ≤≤ ≤≤ ≥ ≤≤× ≤ ≤ ≥ ≤≤ ≤≤× ×≤ ≥ ×≤ ×≤≤ ≥ ×≤×≤≤ ≤≤≤× ≥ ≤≤ ≤≤≤ ≤ ≥ ≤ ≤ ≤≤×≤ ≥ ≤ ×× ×≤ ≥ ×≤ ≤ ≤ ≤ ≥ × ≤≤×≤ ≥ ≤≤ ≤≤ ≤ ≥ ≤≤ ≤× ≤≤ ≥ × ≤≤≤ ≤≤× 合计2 24个家系各群体内的遗传距离家系 ≤中父母本之间的遗传距离为 父本对其子代的遗传距离为 母本对其子代的遗传距离为 子代个体间的遗传距离为 家系 ≤≤中父母本之间的遗传距离为 父本对其子代的遗传距离为 母本对其子代的遗传距离为 子代个体间的遗传距离为 家系≤≤ 中父母本之间的遗传距离为 父本对其子代的遗传距离为 母本对其子代的遗传距离为 子代个体间的遗传距离为 家系⁄ 中父母本之间的遗传距离为 父本对其子代的遗传距离为 母本对其子代的遗传距离为 子代个体间的遗传距离为 ∀期张国范等 皱纹盘鲍中国群体和日本群体的自交与杂交ƒ 的 °⁄标记图 个家系的 °⁄扩增情况ƒ × °⁄ ∏≥ 引物对四个家系的扩增产物大多为多态扩增位点数中等 ≥ 引物对四个家系的扩增产物大多为单态扩增位点数很高 引物∀ ) 家系 ≤ 表示母系 表示父系 其余为子代 ) 家系 ≤≤ 表示母系 表示父系 其余为子代 ) 家系≤≤ 表示母系 表示父系 其余为子代 )家系⁄表示母系 表示父系 其余为子代2 3 4个家系各群体内的平均杂合度家系 ≤子代群体的平均杂合度为 家系 ≤≤子代群体的平均杂合度为 家系≤≤ 子代群体的平均杂合度为 家系⁄ 子代群体的平均杂合度为 ∀2 4 4个家系子代群体间的遗传距离个家系子代家系间遗传距离的观察值为 ) 但最大值不是发生在 ≤≤和⁄之间 表 ∀表2 4个家系群体间的遗传距离× × ≤≤≤≤≤⁄≤ ≤≤ ≤≤ ⁄2 5 总群体的基因多样性及基因分化系数个子代总群体的基因多样性为 基因分化系数为 ∀海 洋 与 湖 沼 卷期张国范等 皱纹盘鲍中国群体和日本群体的自交与杂交ƒ 的 °⁄标记3讨论从扩增图片来看 °⁄呈典型的孟德尔共显性遗传规律 虽不能区分某一座位扩增出的⁄ 片段是纯合的还是杂合的 但其探测基因多态性效率高 是研究杂种优势的极好工具 可以从分子水平上提供基因多态性与杂种优势的关系∀个家系的平均杂合度说明两个杂交组合家系的杂合度均高于两个自交组合 从而可以预测杂交可能产生杂种优势 而这其中尤以亲本遗传距离最大的家系 ≤最为突出∀ ≤的两亲本间遗传距离为 家系 ≤≤的两亲本间的遗传距离为 家系≤≤ 的两亲本间遗传距离为 家系⁄ 的两亲本间遗传距离为 ∀说明本文中所用亲本的两不同地理群体的个体间的遗传距离确实大于两同一地理群体的亲本个体间遗传距离 但因样本少所以尚不能定论∀而中国与日本野生亲本的遗传距离都较大的事实说明 皱纹盘鲍的野生资源保护较好∀由亲本间较大的遗传距离可以产生子代群体较大杂合度这一事实说明 亲本间遗传差异大是产生杂种优势的基础∀然而 家系≤≤ 的亲本遗传距离虽较 ≤≤!⁄ 大很多 在家系的杂合度上并未表现出明显的增高 这其中原因尚不能很好说明∀但可以肯定的是 杂种优势是一复杂的遗传现象 亲本间的遗传差异无疑是重要的原因之一∀由表 可以看出 家系⁄ 与 ≤≤的子代群体间的遗传距离小于其他群体间的遗传距离 这其中的原因有可能是由于当初实验设计受实验条件所限造成的系统的部分开放所致∀另一原因可能是这种单交子代群体仅仅由单一个体父母亲本产生 而且各群体个体间的遗传变异水平较高∀因此 由两对个体产生的子代群体的群体间遗传距离能否与其这两个群体的两对父母本的遗传距离有对应的线性关系还值得讨论∀现有的理论只能比较个体与个体或群体与群体间遗传距离 还无法比较一对父母本对另一对父母本之间的遗传距离∀两个纯合子代群体在某一位点的基因频率很可能会同时都很高 导致/两个纯合子代群体间0的 值高于/一个纯合子代群体与一个杂合子代群体间0或/两个杂合子代群体间0的 值 从而造成在两个纯合子代群体间Π值 即群体间遗传距离 最小的事实∀因此 不能简单地用父母本间的遗传距离来推断子代群体间遗传距离∀各家系子代群体与父母亲本的遗传距离表明皱纹盘鲍的子代倾向于父本基因型的几率稍大 即雄性遗传占主导地位 在兴国红鲤Χψπρινυσχαρπιο√ .σινγυονενσισ与德国镜鲤Χψπρινυσχαρπιο√ σπεχυλαρισ的杂交中也出现类似结果 董在杰等 这其中的具体原因尚待进一步研究∀皱纹盘鲍在基因组水平上杂合度提高的贡献权重雄性占优这一结果在理论上有重要意义∀这一结果的应用价值在于 只要引进雄性亲本进行杂交 即有可能获得杂种优势∀皱纹盘鲍的繁殖特点是成熟雌性的产卵量只有几十万到百万 精子量则以数十亿计∀因此 在生产上只要引进雄性亲本并与本地群体杂交即可获得杂种优势 这样可大大降低生产成本 其潜在的经济效益是巨大的∀各家系子代间的遗传距离表明各家系子代个体间的遗传距离都远远低于其野生父母亲本间的遗传距离及子代与亲本的遗传距离 而子代各群体间遗传距离均小于 指示人工繁育过程中 一定要避免用人工培育群体内或群体间的子一代个体作为父母亲本 以免引起种质资源的严重退化 也提醒我们保护好现有的各地理群落的野生资源对鲍养殖业的可持续发展是非常重要的∀海洋与湖沼 卷从本文中的实验结果可以看出 直接利用中国野生皱纹盘鲍与日本野生皱纹盘鲍杂交可能获得较好杂种优势 本文为此提供了充分的分子生物学证据 同时也说明 °⁄做为一种简单快捷的分子标记 是预测杂种优势的极好手段 可以广泛应用于分子标记辅助育种∀但本文中提供的分子生物学证据也仅限于提供了杂种优势产生的趋势和标记∀由于杂种优势遗传机理极其复杂 一个杂交组合能否表现杂种优势不仅与双亲遗传差异有关 还与双亲遗传背景!所携带等位基因的类型!基因效应种类以及环境等一系列因素有关 许明辉 ∀杂交子代在生产上能否产生某些优良性状以及这些性状能否与这种标记相对应尚需做进一步研究∀致谢在实验过程中得到中国水产科学研究院黑龙江水产研究所孙孝文研究员!梁利群副研究员 以及中国科学院海洋研究所宋林生研究员和刘保忠博士的热心指导 谨致谢忱∀参考文献石拓 孔杰 刘萍等 中国对虾遗传多样性的 °⁄分析)))朝鲜半岛西海岸群体的⁄ 多态性 海洋与湖沼 )吕雪梅 杨关福 张细权等 蛋鸡品系 °⁄变异及其与杂种优势关系的分析 遗传 )刘萍 孔杰 石拓等 中国对虾黄渤海沿岸群亲本及子一代 °⁄分析 海洋水产研究 ) 刘必谦 戴继勋 巨蛎属牡蛎遗传多样性研究 水产学报 )刘必谦 戴继勋 喻子牛 °⁄标记在大连湾牡蛎种群研究中的应用 青岛海洋大学学报 )许明辉 烟草数量性状遗传距离与杂种优势关系的研究 遗传 )孙易 宋文芹 钟贻诚等 用 °⁄和 ƒ °的方法对中国卤虫 种及亲缘关系的研究 遗传学报 )孙效文 梁利群 鲤鱼的遗传连锁图谱 初报 中国水产科学 )宋林生 相建海 周岭华等 六种海产虾类基因组⁄ 多态性的 °⁄标记研究 海洋与湖沼 ) 宋林生 相建海 李晨曦等 日本对虾野生种群和养殖种群遗传结构的 °⁄标记研究 海洋与湖沼 )李斌 鲁成 周泽扬等 °⁄标记构建家蚕分子连锁图 遗传学报 )李祥龙 田庆义 马国强等 波尔山羊杂交后代及其亲本随机扩增多态⁄ 研究 遗传 )孙致良 张超良 金德敏等 °⁄技术在玉米自交系亲缘关系研究中的应用 遗传学报 )张细权 李加权 杨关福 动物遗传标记 北京 中国农业大学出版社 ) )张国范 李霞 我国贝类大规模死亡的现状 中国水产 )张四明 邓怀 晏勇等 中华鲟随机扩增多态性⁄ 及遗传多样性研究 海洋与湖沼 )季维志 宿兵 遗传多样性研究的原理与方法 杭州 浙江科学技术出版社 )梁利群 孙效文 闫学春 °⁄技术分析荷包红鲤抗寒品系与亲本的基因组变化 中国水产科学 ) 滕春波 孙效文 沈俊宝等 利用异源精子激发雌核发育的银鲫及亲本的 °⁄分析 水产学报 ) 董在杰 夏德全 吴婷婷等 兴国红鲤和散鳞镜鲤杂种优势的 °⁄分析 上海水产大学学报 )萨姆布鲁克 弗里奇∞ƒ 曼尼阿蒂斯×著 金冬雁 黎孟枫 张励等译 分子克隆 北京 科学出版社 )° × ⁄ √ √ ≤ Χρασσοστρεαϖιρ2γινιχα )∏ √ ≤ 2 ° ⁄ √ √ • 2 ) ⁄ ⁄⁄ εταλ ° ∏ Πεχτενµαξµιυσ • √ ⁄ ∞ ° ≥ )期张国范等 皱纹盘鲍中国群体和日本群体的自交与杂交ƒ 的 °⁄标记× ⁄εταλ ⁄ Τετραηψµενατηερµοπηιλα )≠ ∂ ∏ ∏¬≠ ⁄ ∞εταλ, ×≤ ≤ × ∞∏ 2 Οστρεαεδυλισ )• ∏ ∏ ° ≥ )∞ • ∏ ∞ ° εταλ ƒ ° √ ∏ 2 Πατινοπεχτενψεσσοενσισ ≥° ∞ ≤ εταλ × ∏ °⁄ ∏ Πλαχοπεχτενµαγελλανιχυσ ≥ )≤ √ ≤ ∏ ∏ ∏ ∏ ⁄ 2 Χρασσοστρεαϖιργινιχα )ΤΗΕΡΑΠ∆ΜΑΡΚΕΡΟΦΣΕΛΦ−ΒΡΕ∆ΑΝ∆ΗΨΒΡΙ∆ΠΡΟΓΕΝΨΒΕΤΩΕΕΝΧΗΙΝΕΣΕΑΝ∆ϑΑΠΑΝΕΣΕΠΟΠΥΛΑΤΙΟΝΣΟΦΗΑΛΙΟΤΙΣ∆ΙΣΧΥΣΗΑΝΝΑΙΙΝΟ∏ 2ƒ • 2 2∞ ± ∞ ∏ 2≠ ÷(ΙνστιτυτεοφΟχεανολογψ,ΤηεΧηινεσεΑχαδεµψοφΣχιενχεσ,Θινγδαο, )(∆αλιανΦισηεριεσΥνιϖερσιτψ,∆αλιαν, )(∆αλιανΙνστιτυτεοφΦισηεριεσΡεσεαρχη,∆αλιαν, )Αβστραχτ× ⁄ °⁄ ∏ ∏ √ 2 Ηαλιοτισδισχυσηανναι ∏ ≤ × ≤ α≅≤ ⎯ ≤≤ ≤ α≅≤ ⎯ ≤≤ ≤ α≅ ⎯ ⁄ α≅ ⎯ × √ ∏ ≤ ≤≤ ≤≤ ⁄ ∏ ∏ 2 ∏ × ≤ ≤≤ ≤≤ ⁄ √ ∏ 2 ∏ ∏ 2 ∏ √ ∏ ≤ ∏ √ × 2 ∏ ≤ ≤≤ ≤≤ ⁄ 2 × ∏ ≤ ≤≤ ≤≤ ⁄ × √ ∏ ¬ √ ∏ΚεψωορδσΗαλιοτισδισχυσηανναι ⁄ ∏ °⁄。

中国人种基因图谱随着分子人类学数据的不断积累,父系Y染色体与母系mtDNA研究的一系列进展,使得中国人群的多样性结构逐渐明晰。
现有的Y染色体数据揭示,现代人出非洲后由东南亚经多次迁徙进入东亚。
在旧石器时代,现代人最初定居东亚或东南亚之后,紧接着不断北迁,这奠定了中国人遗传结构的基础。
通过了解人类基因的遗传成分,绘制中国人种基因图谱。
中国人种基因图谱:一对为性染色体,XY组合的为男性,XX组合的为女性。
Y染色体只在父亲与儿子传代,呈严格的父系遗传,研究Y染色体,可以发现人群在父系关系上的迁徙和发展。
母系mtDNA表现为母系遗传。
通过检测现代人mtDNA,能弄清各民族、各地人的母系血缘关系。
通过研究Y-DNA与mtDNA的重合型,可以揭开中国人的祖先来源之谜。
单倍群人类父系基因Y-DNA人类母系基因mtDNAY-DNA与mtDNA的重合型中国人起源华夏族的起源与形成单倍群在分子进化的研究中,单倍群或单倍型类群是一组类似的单倍型,它们有一个共同的单核苷酸多态性祖先。
因为单倍群由相似的单倍型组成, 所以可以从单倍型来预测单倍群.单核苷酸多态性试验被用来确认单倍型。
单倍群以字母来标记,并且以数字和一些字母来做补充,,例如O3a4。
Y染色体和线粒体单倍群有不同的单倍群标记方法。
单倍群用来标记数千年前的祖先来源。
在人类遗传学中, 最普遍被研究的单倍群是『人类Y染色体脱氧核糖核酸单倍群(Y-DNA单倍群)』和『人类线粒体脱氧核糖核酸单倍群(mtDNA单倍群)』,这两个都可以被用来定义遗传群体。
Y染色体脱氧核糖核酸单倍群仅仅被从父系线遗传,同时mtDNA仅仅被从母系线遗传。
人类父系基因Y-DNA在人类基因学里,人类Y染色体DNA单倍型类群通过Y染色体遗传变异特性进行人类学研究的一门科学,主要用于研究人类的“非洲起源论”及以后的种群分布的遗传学证据。
人类有23对46条染色体,其中22对44条为常染色体,另外一对为性染色体,XY组合的为男性,XX组合的为女性。
DOI : 10.3969/j.issn.l672-9463.2021.05.001•论著.基于综合数据库分析ASXL1基因突变对 子宫内膜癌预后的影响及作用机制范明1韩馨乐$杜俊彳【摘要】目的 通过多个肿瘤综合数据库分析ASXL1基因突变对子宫内膜癌(EC)预后的影响及其作用机制。
方法 全面检索 GEPIA 、Kaplan-Meier plotter 、the Human Protein Atlas 、FireBrowse 、STRING 、UALCAN 等数据库,分析其生存曲线、功能富集情况、基因相关性及信号通路,并进行生物信息学预测。
结果ASXL1基因突变是EC 不良预后因 素,对总生存期及无复发生存期均产生影响(PO.001 )。
相关性分析显示,ASXL1基因突变可能通过表观遗传学,JAK2/STAT3和PI3K/AKT/mT0R 信号通路发挥作用;结合功能富集分析,ASXL1突变主要影响PI3K/AKT/mTOR 信号通路。
结论ASXL1基因突变是EC 患者的不良预后因素,可能通过PI3K/AKT/mTOR 信号通路发挥作用,应进一步开展相关生物学研究,并探索PI3K/mT0R 等信号通路抑制剂的临床价值。
[关键词]子宫内膜癌ASXL1基因突变预后作用机制Prognostic effect and mechanism of A SXL1 gene mutation in endometrial carcinoma based on comprehensive database FanYue, HanJGnle, Du Jun. Guangzhou Women and Children Medical Care Center, G uangzhou 510623[Abstract] Objective To analyze the prognostic effect and mechanism of ASXL1 gene mutation in endometrialcarcinoma (EC) through multiple comprehensive tumor databases. Methods A comprehensive search of GEPIA, Kaplan-Meier plotter, the Human Protein Atlas, FireBrowse, STRING, UALCAN databases, analysis of survival curves,functional enrichment, gene correlation and signal pathways were conducted, with bioinformatics prediction. ResultsASXL1 gene mutation was a poor prognostic factor of EC, affecting both overall survival and relapse —free survival (P<0.001). Correlation analysis showed that ASXL1 gene mutation might play a role through epigenetics, JAK2/STAT3 and PI3K/A.KT/mT0R signaling pathways; combining with functional enrichment analysis, we found that ASXL1 mutation mainly affected PI3K/AKT/mTOR signaling pathway. Conclusion ASXL1 gene mutation is a poor prognosticfactor in EC patients, and may play a role through the PI3KZA.KT/mTOR signaling pathway. Further related biological research should be carried out and the clinical value of PI3K/mT0R inhibitors should be explored.[Key words] Endometrial carcinoma ASXL1 Gene mutation Prognosis Mechanism子宫 内膜癌(Endometrial carcinoma , EC )是女 性第六大常见的恶性肿瘤。
中日韩人种基因拷贝数变异图谱出炉韩国首尔大学基因医学研讨所徐廷瑄教授指导的研讨小组宣称,他们经过对30名中国人、韩国人和日自己的基因组研讨,成功绘制出中日韩人种超高清基因拷贝数变异图谱,并依据该图谱发现,亚洲人独有的基因拷贝数变异共有3500多个。
所谓基因拷贝数变异(Copy Number Vriations)是指在人类基因组中普遍存在的,从1000bp(碱基对)到数百万bp范围内的缺失、拔出、重复和复杂多位点的变异。
研讨说明,不少人类复杂性状疾病都和拷贝数变异有亲密关系。
2021年,第一张人类基因组第一代基因拷贝数变异图谱问世。
这张遗传图谱是经过对欧洲、非洲和亚洲祖先4团体群的270个集体样品停止剖析,用两个互补的技术——单核苷酸多态性(SNPs)基因分型和以克隆为基础的比拟基因组杂交停止基因拷贝数变异挑选,取得了一共1447个拷贝数变异。
之后的一系列研讨显示,基因拷贝数变异是集体之间在基因组序列差异上的一个重要源泉,是研讨基因组退化和表型差异的一个重要要素。
许多关于基因拷贝数变异的研讨结果说明,拷贝数变异可招致不同水平的基因表达差异,对正常表型的构成及疾病的发作开展具有一定作用。
拷贝数变异研讨在法医学方面也具有重要意义,在探求法医学集体识别的遗传变异时不能疏忽拷贝数变异这一基因组多样性的新方式。
首尔大学医学院此次绘制的基因拷贝数变异图谱与西方绘制的现有图谱不同,是只针对中日韩人种停止研讨并绘制完成的,将有效适用于特定人群的疾病诊疗,并为今后正式研讨基因拷贝数变异和疾病之间的关联性提供了良好平台。
(薛严)当第一张人类基因组草图问世时,我们对这一划时代的成就充溢等候,盼望它在医学诊断、预防和治疗方面,可以迅速兑现基因组研讨的初衷。
10年过去了,我们发现那不过是生命迷信这部天书的扉页。
基因组测序现已不算难事,迷信家面临的更大应战,是从浩繁的基因组序列中找到惠及安康的有用信息。
或许,研讨基因拷贝数变异,我们才翻到了这部天书的某一章节。
全球基因图曝光,汉族人竟与日本人基因成分相似?
之前,美国著名的生物学专家们对于全世界各地的人的基因做了一个系统的研究和实验,揭开了现代人基因来源,希望通过生物学的角度由此揭开那些有争议的人种来源,让历史的事实得到最科学的解释。
从这张图中,我们可以看到一个国家人种的纯度。
首先来看看我们国家的汉人、蒙古和西藏人的基因来源
基本上从这这上面我们可以看到的汉人的百分之70以上的基因来源还是我们自己,无论是南方还是北方,差异很微妙。
我国的蒙古人和西藏人的基因自身流传的基因还是占了很大的部分。
但是我们看看日本的就会发现,日本人的基因和我们汉族有着很相似的地方.东亚—南亚的基因都有着很高的比例。
从上面的图片我们也看的处各国的一些基因分布,在这就不做解释了,大家可以自行观看。
中日韩人种基因拷贝数变异图谱出炉
韩国首尔大学基因医学研究所徐廷瑄教授领导的研究小组宣称,他们通过对30名中国人、韩国人和日本人的基因组研究,成功绘制出中日韩人种超高清基因拷贝数变异图谱,并根据该图谱发现,亚洲人独有的基因拷贝数变异共有3500多个。
所谓基因拷贝数变异(Copy Number Vriations)是指在人类基因组中广泛存在的,从1000bp(碱基对)到数百万bp范围内的缺失、插入、重复和复杂多位点的变异。
研究表明,不少人类复杂性状疾病都和拷贝数变异有密切关系。
2019年,第一张人类基因组第一代基因拷贝数变异图谱问世。
这张遗传图谱是通过对欧洲、非洲和亚洲祖先4个人群的270个个体样品进行分析,用两个互补的技术——单核苷酸多态性(SNPs)基因分型和以克隆为基础的比较基因组杂交进行基因拷贝数变异筛选,获得了一共1447个拷贝数变异。
之后的一系列研究显示,基因拷贝数变异是个体之间在基因组序列差异上的一个重要源泉,是研究基因组进化和表型差异的一个重要因素。
许多关于基因拷贝数变异的研究结果表明,拷贝数变异可导致不同程度的基因表达差异,对正常表型的构成及疾病的发生发展具有一定作用。
拷贝数变异研究在法医学方面也具有重要意义,在探索法医学个体识别的遗
传变异时不能忽略拷贝数变异这一基因组多样性的新形式。
首尔大学医学院此次绘制的基因拷贝数变异图谱与西方绘制的现有图谱不同,是只针对中日韩人种进行研究并绘制完成的,将有效适用于特定人群的疾病诊疗,并为今后正式研究基因拷贝数变异和疾病之间的关联性提供了良好平台。
(薛严)
当第一张人类基因组草图问世时,我们对这一划时代的成就充满期待,渴望它在医学诊断、预防和治疗方面,能够迅速兑现基因组研究的初衷。
10年过去了,我们发现那不过是生命科学这部天书的扉页。
基因组测序现已不算难事,科学家面临的更大挑战,是从浩繁的基因组序列中找到惠及健康的有用信息。
或许,研究基因拷贝数变异,我们才翻到了这部天书的某一章节。