拷贝数变异
- 格式:ppt
- 大小:1.37 MB
- 文档页数:27

肿瘤驱动基因的特征和功能研究肿瘤驱动基因是引发癌症的一类基因,它们可以在细胞内发挥重要的功能,促进癌症的发展和生长。
随着分子生物学的研究,我们已经了解了许多肿瘤驱动基因的特征和功能。
本文将探究肿瘤驱动基因的定义、特征及其功能,以期更好地理解肿瘤病理生理学。
1. 肿瘤驱动基因的定义肿瘤驱动基因,即促进细胞癌变和肿瘤形成的基因,这些基因有不同的作用,但它们的共同点是能够通过突变、拷贝数增加等方式对细胞的生长和分裂产生重要影响。
肿瘤驱动基因的存在是一个重要的发现,它引起了关于癌症起源和发展的许多理论。
2. 肿瘤驱动基因的特征肿瘤驱动基因的特征主要体现在以下几个方面:(1)拷贝数变异:肿瘤驱动基因的拷贝数改变可以是整倍体增加也可以是基因座突变。
这种变异可能唤起肿瘤形成。
(2)突变:某些突变会导致肿瘤抑制基因失去了正常功能,同时肿瘤驱动基因的突变往往会导致基因失去控制。
(3)化学修饰:一些化学修饰如DNA甲基化、羟甲基化等也可能影响肿瘤驱动基因的功能。
3. 肿瘤驱动基因的功能肿瘤驱动基因可以对细胞的正常生理功能产生影响,并促进癌症的发展。
其主要功能体现在三个方面:(1)参与细胞生长和分裂的调节:肿瘤驱动基因可以促进或抑制细胞的生长和分裂,这是它们的基本功能。
突变或拷贝数增加等变异可能导致基因失去对生长和分裂的正常调节作用,从而引起癌细胞的不受限制生长。
(2)细胞信号通路的激活:细胞通路是细胞间通信网络的重要组成部分,肿瘤驱动基因可以通过激活信号通路的某些分子来参与信号传递。
这些分子可能是细胞内的激活酶、受体或转录因子等,在突变时导致某些分子一直处于激活状态。
(3)参与细胞程序化死亡调节:细胞程序性死亡(apoptosis)是细胞生命周期的一个重要环节,避免细胞癌变的发生。
但在某些情况下,肿瘤驱动基因可以抑制细胞凋亡的发生,从而促进持续的生长。
这种抑制可能与直接抑制凋亡信号通路的分子有关,或者与促进细胞生长和分裂的信号通路紊乱有关。


不可不看的CNV全解析导读拷贝数变异(Copy number variation,CNV)是由基因组发生重排而导致的,一般指长度为1kb以上的基因组大片段拷贝数的增加或减少,主要表现为亚显微水平的缺失或重复。
这种变异既有个体的正常多态性变异,也有致病性的变异[1]。
目前,按照CNV是否致病可分为致病性CNV、非致病性CNV和不明临床意义CNV。
对于如何解读检测出的CNV临床意义,长期困扰着临床医生。
1拷贝数变异概念拷贝数目变异也称拷贝数目多态,是一种长度大于1kb的DNA片段的变异,在人类基因组中广泛分布,CNV位点的突变率远高于SNP (Single nucleotide polymorphism),是人类疾病的重要致病因素之一。
2检测方法目前全基因组检测方法主要有微阵列比较基因组杂交(Array CGH)和最新的基于高通量测序技术的染色体异常检测(NGS染色体异常检测)。
两种技术相比较:由于Array CGH是在已知探针前提下进行检测,所以无法检测出未知的CNV;成本偏高,受检者负担较重;而NGS染色体异常检测为最新的CNV检测技术,对样本要求较低,能发现更多的新变异CNV,在国内临床已得到广泛应用。
关于检测出CNV的遗传咨询,是目前临床医生的一个难点,特别是对于不明临床意义的CNV。
希望通过以下两篇关于CNV解读的文章为临床医生的遗传咨询提供参考,以便更好的与患者及其家属沟通。
3CNV解读流程(1)欧洲人类遗传学杂志发表的一篇关于CNV解读流程的文章[2],该文章把CNV解读过程分为五步,以此进行CNV解读。
图1 CNV解读流程[2]第一步:得到CNV结果,与序列数据库进行比对,能比对到数据库中,人群分别频率n≥1%,说明CNV为人群中比较常见多态性即非致病性CNV。
对于没有比对到数据库中的结果进行下一步;第二步:与基因组变异数据库(Database of Genomic Variants)比对,数据库中出现正常次数n≥3,表明CNV为正常多态性即非致病性CNV。

拷贝数变异(CNV)的概念和影响拷贝数变异(CNV)是指基因组中在一些个体中重复或缺失的DNA片段,它们通常大于1 kb,可以涉及一个或多个基因。
CNV是一种常见的基因组变异,它们在人类基因组中占据约12%的区域,影响约4400个基因。
CNV可以通过不同的机制产生,如不对称的同源重组、非同源末端连接、转座等。
CNV可以影响基因的表达水平、功能和相互作用,从而导致不同的表型和性状。
CNV与许多人类疾病有关,如癌症、神经退行性疾病、自闭症等。
CNV的检测方法和挑战CNV的检测方法主要有两类:基于芯片的方法和基于测序的方法。
基于芯片的方法是利用微阵列芯片或SNP芯片对基因组进行杂交分析,根据信号强度的变化推断CNV的存在与否。
基于测序的方法是利用高通量测序技术对基因组进行测序分析,根据覆盖度或连接信息推断CNV 的位置和大小。
CNV的检测方法面临着一些挑战,如:•基于芯片的方法只能检测到比较大的CNV(>10 kb),而且受到芯片设计和分辨率的限制。
•基于测序的方法需要大量的计算资源和复杂的算法,而且受到测序深度和质量的影响。
•不同方法之间存在一定的差异和不一致,需要进行标准化和整合。
•CNV与性状之间的关联分析需要考虑多种因素,如遗传背景、环境因素、表观遗传修饰等。
CNV在英国生物数据库中的新发现在一项新的研究中,来自美国布罗德研究所、布莱根妇女医院和哈佛医学院的研究人员开发出一种计算方法,在英国生物数据库(UK Biobank)中检测到1500万个CNV,比以前对相同数据的分析结果多出六倍。
英国生物数据库是一个包含了50万名志愿者的健康和遗传信息的大型数据库,它为研究人员提供了一个研究人类性状和疾病风险的宝贵资源。
研究人员使用了一种名为cnv-scan(copy-number variant scan)的计算方法,它可以利用英国生物数据库中已有的SNP芯片数据来检测CNV。
cnv-scan方法具有以下几个特点:•它可以检测到比较小的CNV(<10 kb),并且可以区分单拷贝变异(SCN)和多拷贝变异(MCN)。

基因的拷贝数
基因的拷贝数是指某个特定基因在染色体上的重复数目。
在不同的物种中,基因的拷贝数可以相同,也可以不同。
人类基因组中有许多基因是拷贝多次的,这些称为基因家族,如血型决定基因家族、组蛋白H2A基因家族等。
基因的拷贝数变异是基因组的一种重要特征,它对人类个体的生长发育、代谢、免疫应答等生理过程都有影响。
此外,不同的基因拷贝数变异与身体状况、疾病易感性和治疗反应等方面也有关联。
因此,基因拷贝数变异在个体基因学和人类遗传学研究中具有重要意义。
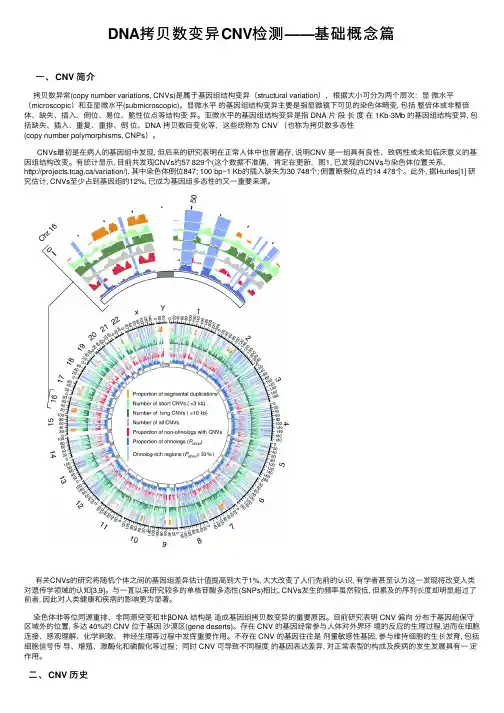
DNA拷贝数变异CNV检测——基础概念篇⼀、CNV 简介拷贝数异常(copy number variations, CNVs)是属于基因组结构变异(structural variation),根据⼤⼩可分为两个层次:显微⽔平(microscopic)和亚显微⽔平(submicroscopic)。
显微⽔平的基因组结构变异主要是指显微镜下可见的染⾊体畸变, 包括整倍体或⾮整倍体、缺失、插⼊、倒位、易位、脆性位点等结构变异。
亚微⽔平的基因组结构变异是指 DNA ⽚段长度在 1Kb-3Mb 的基因组结构变异, 包括缺失、插⼊、重复、重排、倒位、DNA 拷贝数⽬变化等,这些统称为 CNV (也称为拷贝数多态性(copy number polymorphisms, CNPs)。
CNVs最初是在病⼈的基因组中发现, 但后来的研究表明在正常⼈体中也普遍存, 说明CNV 是⼀组具有良性、致病性或未知临床意义的基因组结构改变。
有统计显⽰, ⽬前共发现CNVs约57 829个(这个数据不准确,肯定在更新,图1, 已发现的CNVs与染⾊体位置关系,http://projects.tcag.ca/variation/), 其中染⾊体倒位847; 100 bp~1 Kb的插⼊缺失为30 748个; 倒置断裂位点约14 478个。
此外, 据Hurles[1] 研究估计, CNVs⾄少占到基因组的12%, 已成为基因组多态性的⼜⼀重要来源。
有关CNVs的研究将随机个体之间的基因组差异估计值提⾼到⼤于1%, ⼤⼤改变了⼈们先前的认识, 有学者甚⾄认为这⼀发现将改变⼈类对遗传学领域的认知[3,9]。
与⼀直以来研究较多的单核苷酸多态性(SNPs)相⽐, CNVs发⽣的频率虽然较低, 但累及的序列长度却明显超过了前者, 因此对⼈类健康和疾病的影响更为显著。
染⾊体⾮等位同源重排、⾮同源突变和⾮βDNA 结构是造成基因组拷贝数变异的重要原因。

copy-number alterations -回复什么是拷贝数变异(copy number alterations)?拷贝数变异是一种常见的基因组变异形式,指基因组上某一区域的DNA 重复次数发生改变。
正常情况下,一个细胞中某一特定基因区域的DNA 序列存在于一定数量的拷贝中。
然而,由于各种原因,这些拷贝数可能会发生变异,导致一个或多个基因区域的拷贝数增加或减少。
这种变异可以出现在染色体的整个区域,也可以是一个基因的局部变异。
拷贝数变异在个体间具有很大的差异,并且已经被证明与多种疾病的发生和发展密切相关。
拷贝数变异的机制是什么?拷贝数变异的机制包括多种可能的事件,如基因重组、突变、插入、缺失、重复和复合等。
这些事件可以导致重复的DNA序列插入到基因组中,或导致基因组中的某一部分丢失。
其中最常见的机制是非同源重组和复制错误。
非同源重组是指两个不同拷贝之间的DNA片段互相重组,导致拷贝数变异。
而复制错误则是指DNA在复制过程中发生错误,如重复插入或缺失某一片段。
拷贝数变异的检测方法有哪些?目前,有多种方法可用于检测拷贝数变异,包括来自基因组学和分子生物学领域的方法。
其中一种常用的方法是比较基因组杂交(Comparative Genomic Hybridization, CGH)。
CGH使用一个探针来检测某个特定基因或区域的DNA序列拷贝数变异。
这种方法通过比较样本中的DNA与参考DNA的杂交信号强度,来确定拷贝数变异的存在与程度。
另一种常用的方法是荧光原位杂交(Fluorescence In Situ Hybridization, FISH),它可以直接观察和检测细胞核中某一特定基因或区域的拷贝数变异。
此外,还可以利用高通量测序技术,如全基因组测序(Whole Genome Sequencing, WGS)或下一代测序(Next Generation Sequencing, NGS)来进行全面、高效的拷贝数变异分析。

拷贝数变异检测方法拷贝数变异是指基因组中某一段DNA序列在进化过程中发生了拷贝数的变异,即该序列的拷贝数增加或减少。
拷贝数变异被认为是基因组结构变异的主要形式之一,它在物种进化和个体遗传多样性中起到重要的作用。
为了准确、高效地检测拷贝数变异,科学家们开发了一系列方法。
下面将介绍几种常用的拷贝数变异检测方法。
1. MLPA(Multiplex Ligation-dependent Probe Amplification)MLPA是一种常用的拷贝数变异检测方法,它利用多重连接依赖式探针扩增技术,可以同时检测多个目标序列的拷贝数。
该方法通过引入两个特异性的引物,使目标序列的两个相邻区域连接起来,然后进行PCR扩增。
通过比较目标序列与参考基因组的扩增产物的相对强度,可以确定目标序列的拷贝数是否发生变异。
2. qPCR(Quantitative Polymerase Chain Reaction)qPCR是一种基于聚合酶链反应的拷贝数变异检测方法,它可以快速、准确地测量目标序列的拷贝数。
该方法利用特异性引物和荧光探针,通过监测PCR反应体系中的荧光信号强度来定量目标序列的拷贝数。
相比于传统PCR方法,qPCR具有更高的灵敏度和准确性。
3. MLST(Multilocus Sequence Typing)MLST是一种基于多基因序列分型的拷贝数变异检测方法,它通过测定多个基因的拷贝数变异来推断目标序列的拷贝数。
该方法利用PCR扩增多个基因的片段,并对扩增产物进行测序分析。
通过比较目标序列与参考基因组的片段长度和序列差异,可以确定目标序列的拷贝数是否发生变异。
4. aCGH(array Comparative Genomic Hybridization)aCGH是一种基于基因组DNA杂交的拷贝数变异检测方法,它可以全基因组范围内快速、高通量地检测拷贝数变异。
该方法利用两个不同来源的DNA样品,将其分别标记为红色和绿色,并将它们杂交到DNA芯片上。

拷贝数变异名词解释
拷贝数变异是指在基因组中存在多个拷贝数不同的基因或
DNA序列。
拷贝数是指一个基因或DNA序列在某个基因组中的重复次数。
拷贝数变异可以是正常人群中的一种常见现象,也可以是导致遗传疾病的原因之一。
在正常情况下,基因组中的某些基因或DNA序列会存在多个
拷贝,这被认为是基因组进化的结果。
这些多个拷贝可能具有不同的功能或表达模式,从而为生物个体提供更多的遗传变异性。
然而,当某个基因或DNA序列的拷贝数发生异常变化时,就可能导致疾病或其他健康问题。
拷贝数变异可能呈现多种形式,包括基因缺失、重复、扩增等。
例如,当某个基因的拷贝数减少时,可能导致该基因的功能丧失或减弱,进而导致相关疾病的发生。
相反,当某个基因的拷贝数增加时,可能导致该基因的过度表达或功能改变,也可能引发疾病。
拷贝数变异的检测和研究对于理解遗传疾病的发病机制和个体差异具有重要意义。
近年来,随着高通量测序技术的发展,拷贝数变异的检测已经成为基因组研究的重要内容之一。
通过对拷贝数变异的分析,可以揭示基因组结构的变异和进化过程,也可以为疾病的诊断和治疗提供有价值的信息。

拷贝数变异及其研究进展摘要:拷贝数变异(Copy number variations, CNVs)主要指1kb-1Mb的DNA片段的缺失、插入、重复等。
文章主要介绍了CNVs的基本知识及其机理,着重介绍了其各种检测技术,并进一步阐明CNVs对人类疾病及哺乳动物疾病的影响。
此外,对其研究发展进行可行性展望。
关键词:拷贝数变异机理检测技术疾病2004年,两个独立实验小组几乎同时报道,在人类基因组中广泛存在DNA片段大小从1 kb到几个Mb范围内的拷贝数变异(CNVs)现象。
在2006 年的《Nature》杂志上,来自英国Wellcome Sanger研究所以及美国Affymetrk公司等多国研究人员组成的研究小组公布了第1张人类基因组的第1代CNV图谱,后续又有3篇文章陆续发表在《Nature Genetics》和《Genome Research》杂志上,聚焦这一重大发现。
受到检测手段的限制,这类遗传变异直到最近2年才为研究者所重视,并迅速成为当前人类遗传学研究的热点。
CNVs 最初在患者的基因组中发现,但后来发现CNVs也大量存在于正常个体的基因组内,主要引起基因(或部分基因)的缺失或增多。
拷贝数的变异过程既与疾病相关,也与基因组自身的进化有关。
针对CNVs的发现,美国遗传学家JamesR.Lupski提出“我们不能再将人与人之间的差异想当然地认为仅是单碱基突变的结果,因为还存在更复杂的来自于CNVs的结构性差异”。
Lupski认为,CNVs的发现将改变人类对遗传学领域的认知,并将影响19世纪被誉为“遗传学之父”的孟德尔及 1953年发现“DNA双螺旋”的弗兰西斯•克里克与吉姆•沃特森所确立的人类遗传学基准1 CNV概述1.1 CNV的概念基因组变异包括多种形式,包括SNPs,数目可变串联重复位点VNTRs (微卫星等),转座元件 (Alu序列等),结构变异(重复、缺失、插入等)。
CNVs指大小从1kb到1Mb 范围内亚微观片段拷贝数突变,这些拷贝片段的缺失、复制、倒置等的变异都统称为CNVs,但不包括由转座子的插人和缺失引起的基因变异(如0-6kb Kpn I重复)[1]。
16号染色体发生重复拷贝数为3的原因16号染色体的重复是一种染色体异常,称为16p11.2重复综合征(16p11.2 duplication syndrome)。
该综合征是一个常见的常染色体显性遗传病,与多种身体和神经行为异常相关。
目前已有多项研究对该综合征的拷贝数为3的原因进行了探究。
拷贝数变异(Copy Number Variations,CNVs)是遗传学中常见的遗传变异形式,指的是其中一特定段的染色体序列在个体之间存在拷贝数量差异。
拷贝数变异在人类基因组中普遍存在,被认为是遗传和个体差异的重要因素。
16p11.2重复综合征的发生是由于该基因区域发生了增加,导致个体中16号染色体的该区域被重复了一次。
目前的研究表明,16p11.2重复综合征的发生与两个主要机制有关:基因复制与染色体不稳定性。
首先,基因复制是16p11.2重复综合征发生的一个重要机制。
16p11.2基因区域包含了多个基因,其中一些被认为在神经系统的发育和功能中起到重要作用。
在染色体复制过程中,染色体的一个片段可能会错误地被复制一次,导致该片段的拷贝数增加。
这种基因复制的错位可能是由于DNA复制过程中的错误、基因重组或其他DNA修复机制的错误引起的。
其次,染色体不稳定性也是16p11.2重复综合征发生的机制之一、染色体在复制和维护过程中很容易出现错误,包括错配、插入、缺失和重排等。
这些染色体不稳定性的错误可能导致整个16号染色体的染色体片段在复制过程中被重复或丢失。
有研究发现,16p11.2重复综合征的发生与家族遗传有关。
在一些家族中,16号染色体的16p11.2区域很容易发生基因复制和染色体不稳定性,导致该区域的拷贝数增加。
家族遗传可能与特定的基因突变、染色体结构变异或遗传修饰因子有关。
此外,环境因素也可能对16p11.2重复综合征的发生起到一定的作用。
环境因素包括母体孕期的营养状况、暴露于毒物或致突变物质、生活方式等。
这些环境因素可能引起胚胎发育过程中的基因表达异常,从而导致染色体的不稳定性。
拷贝数变异(CNV)人类基因组由23对染色体中的60亿个碱基(或核苷酸)组成。
正常人类基因组成分通常是以2个拷贝存在,分别来自父母。
拷贝数变异(CNV)是由基因组发生重排而导致的,一般指长度为1kb以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复,是人类疾病的重要致病因素之一。
异常的DNA拷贝数变化(CNV)是许多人类疾病(如癌症、遗传性疾病、心血管疾病)的一种重要分子机制。
作为疾病的一项生物标志,染色体水平的缺失、扩增等变化已成为许多疾病研究的热点,然而传统的方法(比如G显带,FISH,CGH等)存在操作繁琐,分辨率低等问题,难以提供变异区段的具体信息。
CNV,即拷贝数变异,一般指长度为1kb到几个Mb基因组大片段的拷贝数复制、缺失。
CNV被定义为一段至少1kb大小DNA的拷贝数,与具有代表性的参考基因组拷贝数不同。
CNV在基因组中的存在形式主要有以下几种:2条同源染色体拷贝数同时出现缺失;1条同源染色体发生缺失,1条正常;1条同源染色体出现拷贝数重复,另1条正常;1条同源染色体出现缺失,另1条出现拷贝数重复;2条同源染色体同时出现拷贝数重复。
染色体拷贝数变异(CNV)检测:NIPT技术目前医院临床应用的为普通NIPT技术,商业上还有通过增加测序数据的升级版的NIPT产品(可以检测染色体微缺失/微重复和某些单基因病)。
对于NIPT提示的CNV可以分为两种:母源性CNV,就是母亲存在CNV(此时胎儿50%可能存在相同的CNV,50%可能不存在该CNV);第二种,胎儿CNV。
母源性CNV的阳性预测值(PPV)接近100%,因为母源游离DNA占比90%,因此阳性预测值(PPV)很高就不足为奇。
但是不同的检测机构或者有些已发表文献,并不提示母源性CNV。
对于母源性CNV,胎儿无非两种情况,和母亲一样拥有同样的CNV,或不含有该CNV。
在临床咨询中,对于这种来源于母源或父源CNV,如果父母本身没有任何表型,胎儿本身也不存在超声结构异常,我们大多认为偏良性。
高通量测序数据的基因组拷贝数变异[]检测方法综述
刘珍;刘永壮
【期刊名称】《生物信息学》
【年(卷),期】2024(22)1
【摘要】拷贝数变异是指基因组中发生大片段的DNA序列的拷贝数增加或者减少。
根据现有的研究可知,拷贝数变异是多种人类疾病的成因,与其发生与发展机制密切相关。
高通量测序技术的出现为拷贝数变异检测提供了技术支持,在人类疾病研究、临床诊疗等领域,高通量测序技术已经成为主流的拷贝数变异检测技术。
虽然不断有新的基于高通量测序技术的算法和软件被人们开发出来,但是准确率仍然不理想。
本文全面地综述基于高通量测序数据的拷贝数变异检测方法,包括基于reads深度的方法、基于双末端映射的方法、基于拆分read的方法、基于从头拼接的方法以及基于上述4种方法的组合方法,深入探讨了每类不同方法的原理,代表性的软件工具以及每类方法适用的数据以及优缺点等,并展望未来的发展方向。
【总页数】8页(P11-18)
【作者】刘珍;刘永壮
【作者单位】哈尔滨因极科技有限公司;哈尔滨工业大学计算机科学与技术学院【正文语种】中文
【中图分类】TP391
【相关文献】
1.高通量测序技术检测早期自然流产组织染色体非整倍体及拷贝数变异的临床意义
2.一种基于高通量测序的拷贝数变异检测自动化分析解读及报告系统
3.基于高通量测序的拷贝数变异检测技术在产前诊断中的临床应用
4.拷贝数变异测序在先天性心脏病胎儿基因组拷贝数变异筛查中的应用
5.利用全基因组重测序数据检测8个鸭品种基因组拷贝数变异
因版权原因,仅展示原文概要,查看原文内容请购买。
illumina芯片拷贝数变异分析流程Analyzing copy number variations (CNVs) in Illumina microarray data can be a challenging but incredibly informative process. Illumina芯片是一种广泛用于基因组学研究的高通量技术,其数据可以提供基因组中拷贝数变异的信息。
CNVs refer to structural variations in the DNA that involve gains or losses of sections of the genome, and they have been implicated in various human diseases. Illumina microarrays are commonly used to detect and analyze CNVs due to their high resolution and ability to simultaneously assess thousands of genetic markers.One of the first steps in the analysis of CNVs from Illumina microarray data is the pre-processing of raw intensity signals. This involves normalization of the data to correct for systematic variations in intensities across samples, as well as quality control measures to assess the reliability of the data. The goal is to ensure that the data is of high quality and free from technical artifacts that could impact the accuracy of CNV calling. Pre-processing of the data is crucial to obtaining reliable results in downstream analyses.After pre-processing, the next step is CNV calling, which involves identifying regions of the genome that exhibit differences in copy number compared to a reference sample. There are various algorithms available for CNV calling from Illumina microarray data, each with its own strengths and limitations. Commonly used algorithms include PennCNV, QuantiSNP, and Nexus Copy Number. These algorithms use statistical models to assess the likelihood of a CNV at specific genomic loci and provide a measure of confidence in the call.Once CNVs have been called, the next step is to annotate and interpret the results. This involves mapping the identified CNVs to the human genome and determining their potential functional consequences. CNVs can impact gene expression, disrupt gene structures, or alter regulatory regions, so understanding their effects is crucial for linking them to disease phenotypes. Various bioinformatics tools and databases can assist in the annotation of CNVs and provide insights into their biological significance.In addition to data analysis, it is essential to validate identified CNVs using independent experimental methods. This can includequantitative PCR, droplet digital PCR, or fluorescence in situ hybridization to confirm the presence and precise boundaries of the CNVs. Validation is critical to ensure the reliability of the findings and eliminate false positives that may arise from bioinformatics analyses. By combining computational analysis with experimental validation, researchers can confidently characterize CNVs and their implications in various diseases.Overall, analyzing CNVs from Illumina microarray data is a comprehensive and multi-step process that requires a combination of bioinformatics skills, statistical knowledge, and experimental validation. Despite the challenges, the insights gained from studying CNVs can provide valuable information about the genetic basis of diseases and pave the way for precision medicine approaches. Illumina芯片数据中CNVs的分析是一项既具有挑战性又极具信息价值的过程。
拷贝数变异测序
拷贝数变异测序是一种新兴的基因测序技术,它通过复制和变异的过程,可以快速高效地获取生物体的遗传信息。
这项技术的应用范围非常广泛,可以帮助科学家们更好地了解基因组的组成和功能。
在拷贝数变异测序中,首先需要提取待测生物体的DNA样本。
然后,通过一系列的实验步骤,将DNA样本进行复制和变异。
复制过程中,DNA会被不断复制成多个拷贝,从而增加了测序的灵敏度和准确性。
而变异过程则是指在复制过程中,可能会发生一些突变,从而使得复制后的DNA序列与原始DNA序列有所不同。
拷贝数变异测序的原理是基于PCR(聚合酶链式反应)技术的改进。
PCR技术可以通过特定的引物,将DNA序列进行扩增。
而拷贝数变异测序则是在PCR技术的基础上,引入了一些特殊的酶和试剂,以实现DNA的复制和变异。
通过不断地进行复制和变异,科学家们可以获得大量的DNA序列信息,从而揭示生物体的遗传特征和功能。
拷贝数变异测序的应用非常广泛。
一方面,它可以帮助科学家们更好地了解基因组的结构和功能,从而推动基因组学研究的发展。
另一方面,它也可以应用于临床医学领域,帮助医生们更好地诊断和治疗疾病。
例如,在肿瘤学研究中,拷贝数变异测序可以帮助科学家们了解肿瘤细胞的遗传特征,从而为个体化治疗提供依据。
总的来说,拷贝数变异测序是一种非常有前景的基因测序技术。
它
可以帮助我们更好地了解生物体的遗传特征和功能,从而推动生命科学研究的发展。
随着技术的不断进步和应用的不断扩大,相信拷贝数变异测序将在未来的研究和临床实践中发挥越来越重要的作用。