无菌检查法方法验证及操作要点共29页文档
- 格式:ppt
- 大小:3.20 MB
- 文档页数:29

安徽捷众生物化学有限公司无菌检查方法(薄膜过滤法)验证方案文件编号:QY·TS·05·007-00批准日期:年月日实施日期年月日安徽捷众生物化学有限公司无菌检查方法(薄膜过滤法)验证方案目录1.验证目的2. 验证频次3. 验证操作内容与要求3.1. 验证操作试验3.2 试验菌要求3.3.验证方法步骤3.4. 验证准备3.5验证试验操作4.验证结果评价分析5.附件1.验证目的确认所采用的方法适合于供试品的无菌检查。
即确认供试品在该检验量、该检验条件下无抑菌活性,以保证检验结果的准确、可靠及检验方法的完整性。
2. 验证频次2.1. 建立药品的无菌检查法时2.2. 修订检验方法时2.3. 供试品的组分或原检验条件发生改变可能影响检验结果时3. 验证操作内容与要求:3.1. 验证时,按“供试品的无菌检查”的规定要求进行验证操作试验:3.1.1. 对每一试验菌应逐一进行验证。
3.1.2.硫乙醇酸盐流体培养基—金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、生孢梭菌。
3.1.3. 改良马丁琼脂培养基—白色念珠菌、黑曲霉。
3.2试验菌要求:3.2.1验证试验所用的菌株传代次数不得超过5代,并采用适宜的菌种保藏技术,以保证试验菌株的生物学特性。
3.2.2验证试验可在供试品的无菌检查之前或与供试品的无菌检查同时进行。
当同时进行时,若验证试验失败,则供试品的无菌检查结果是不成立的3.3.验证方法步骤:3.3.1验证试验的操作计划用3个不同批号产品按照无菌检查方法(薄膜过滤法)进行平行试验,通过计算回收率来判断无菌检查方法(薄膜过滤法)是否对产品有影响。
3.3.2试验结果可接受标准用无菌标准评价方法“0.9%氯化钠注射液(三层共挤膜袋)的无菌检查”对检品中无菌检查试验结果应显示3次独立的平行试验中,稀释剂对照组的菌回收率应不小于70%,试验组的回收率也不低于70%。
3.4. 验证准备:准备验证所需的供试品、培养基、试药试剂、仪器设备和菌种。
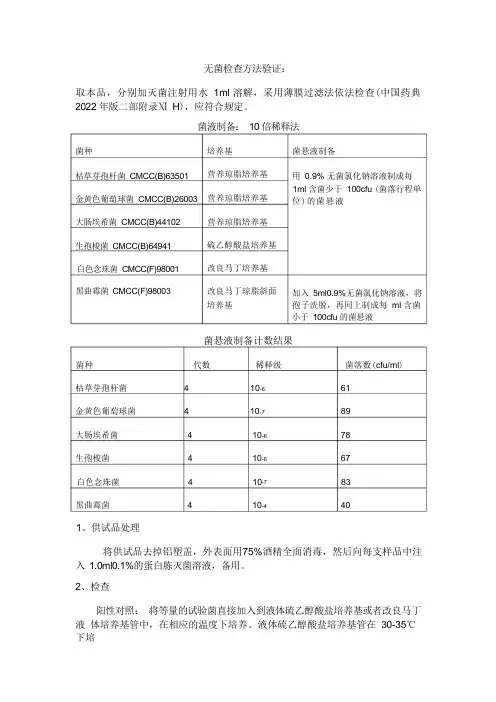
无菌检查方法验证:取本品,分别加灭菌注射用水 1ml 溶解,采用薄膜过滤法依法检查(中国药典 2022 年版二部附录Ⅺ H ),应符合规定。
菌液制备: 10 倍稀释法培养基 营养琼脂培养基营养琼脂培养基营养琼脂培养基硫乙醇酸盐培养基改良马丁培养基改良马丁琼脂斜面 培养基菌悬液制备计数结果菌种 代数 稀释级 菌落数(cfu/ml )枯草芽孢杆菌 4 10-6 61金黄色葡萄球菌 4 10-7 89大肠埃希菌 4 10-6 78生孢梭菌 4 10-6 67白色念珠菌 4 10-7 83黑曲霉菌 4 10-4 401、供试品处理将供试品去掉铝塑盖,外表面用75%酒精全面消毒,然后向每支样品中注 入 1.0ml0.1%的蛋白胨灭菌溶液,备用。
2、检查阳性对照: 将等量的试验菌直接加入到液体硫乙醇酸盐培养基或者改良马丁液 体培养基管中,在相应的温度下培养。
液体硫乙醇酸盐培养基管在 30-35℃下培菌种枯草芽孢杆菌 CMCC(B)63501 金黄色葡萄球菌 CMCC(B)26003 大肠埃希菌 CMCC(B)44102生孢梭菌 CMCC(B)64941白色念珠菌 CMCC(F)98001黑曲霉菌 CMCC(F)98003菌悬液制备用 0.9% 无菌氯化钠溶液制成每 1ml 含菌少于 100cfu (菌落行程单 位)的菌悬液加入 5ml0.9%无菌氯化钠溶液,将 孢子洗脱,再同上制成每 ml 含菌 小于 100cfu 的菌悬液养;改良马丁液体培养基管在23-28℃下培养3-5 天。
观察记录。
阴性对照:将灭菌的液体硫乙醇酸盐培养基或者改良马丁液体培养基管直接放在相应的温度下培养。
液体硫乙醇酸盐培养基管在30-35℃下培养;改良马丁液体培养基管在23-28℃下培养3-5 天。
观察记录。
样品 (薄膜过滤法):每种实验菌取10 支处理好的供试品溶液,将溶液合并后加入制备好的菌悬液1ml,用0.1%的蛋白胨灭菌溶液稀释至100ml,按薄膜过滤法过滤,取出滤膜,将其分为3 等份,分别置于含硫乙醇酸盐流体培养基及改良马丁培养基的容器中,其中一份作为阳性对照用。






⽆菌检验⽅法验证1. ⽬的(PURPOSE)规范药品质量检查⽅法的管理,确保药品所⽤⽆菌检验⽅法的可⾏性、检验结果的可靠性。
2. 适⽤范围(SCOPE)适⽤于药品的⽆菌检验⽅法验证。
3. 职责(RESPONSIBILITY)质量控制部微⽣物组负责4. 关键词(KEY WORDS)⽆5. 定义(DEFINITION)⽆6. 程序(PROCEDURE)6.1 验证的原理6.1.1验证试验⽅案的设计需要满⾜两个⽅⾯的要求:1)在检测前样品的预处理⽅式能有效抵消(或中和)产品中抑菌性;2)样品的预处理⽅式和检测过程以及培养条件等均不会影响样品中的微⽣物⽣长。
6.1.2 对同类产品三个不同批号样品进⾏微⽣物挑战试验后,通过⽐较三批样品中挑战菌株的恢复⽣长结果来评价整个检验⽅法的准确性、有效性和重现性。
6.2 验证试验⽤供试品的制备与检查6.2.1 样品组(A组):⽤适当的⽅法中和样品,再向其中接⼊少量的挑战微⽣物(接种量控制在10~100cfu之间),按照常规检测程序和培养条件检查挑战微⽣物的恢复⽣长情况。
6.2.2 蛋⽩胨对照组(B组):除了⽤A溶液(0.1%蛋⽩胨溶液)替代实际样品外,中和⽅式、检测程序和培养条件等均与样品组(A组)相同。
6.2.3 阳性对照组(C组):不经中和处理,将微⽣物挑战菌株直接接种于培养基培养,接种量与前两组相同,均在10~100cfu 之间。
6.3 验证⽤挑战微⽣物6.3.1⽤于验证试验的挑战微⽣物要具有⼴泛代表性,⾄少包括需⽓菌、厌氧菌、兼性菌等,所选⽤的菌株要基本上涵盖样品中可能存在的各类微⽣物。
在验证中常选⽤的微⽣物有⾦黄⾊葡萄球菌、枯草芽孢杆菌、⼤肠杆菌、铜绿假单胞菌、⽣孢梭菌、⽩⾊念珠菌、⿊曲霉等。
6.3.2 挑战微⽣物的培养条件及贮存条件需按《QC-6.2菌种管理》进⾏,以确保各代微⽣物细胞的⽣理状态保持稳定⼀致。
6.3.3验证时,选择的挑战微⽣物传代次数不应超过5代,并且详细记录所⽤菌株的来源、传代次数。

无菌检查法要点介绍及检验操作关键点培训无菌检查法是一种用来检验物品是否受到微生物污染的方法。
在医疗、食品生产等领域都有广泛的应用。
下面我们就来介绍一下无菌检查法的要点和检验操作关键点。
一、无菌检查法的要点介绍1.原理:无菌检查法主要是通过将待检物品在无菌条件下接种到含有富集培养基的培养基上,然后进行培养观察,确定是否有微生物生长来判断物品是否受到污染。
2.方法:无菌检查主要包括直接接种法和膜过滤法。
直接接种法适用于液体和固体物品的检验,而膜过滤法适用于液体物品的检验。
通过这两种方法可以有效地检验物品是否受到微生物污染。
3.操作流程:进行无菌检查的操作流程一般包括样品的采集、无菌操作台的准备、操作者的衣着换洗、培养基的配置、样品的接种和培养观察等环节。
4.结果判定:通过观察接种后的培养基是否有微生物生长,来判断物品是否受到微生物污染。
根据不同的培养基和生长条件,可以确定是否存在细菌、真菌等微生物。
5.记录和报告:无菌检查的结果需要进行记录和报告,如样品信息、接种情况、培养观察结果等,以便于后续的跟踪和管理。
二、无菌检查法的操作关键点培训1.无菌操作培训:操作者需要接受无菌操作的相关培训,包括穿戴无菌服、洗手消毒、操作台的准备等技能的培训。
2.培养基的准备:操作者需要了解不同类型的培养基的准备方法和要求,包括培养基的pH值、温度、灭菌条件等。
3.样品采集:对于不同类型的样品,采集方法和采集点会有所不同,操作者需要了解样品采集的相关知识和技能。
4.接种方法:无菌操作台的准备、样品的接种和培养基的密封等操作步骤需要掌握,以保证接种的无菌条件。
5.培养观察:操作者需要了解不同微生物的培养观察条件和特征,以便于正确地判断培养基上的微生物生长情况。
6.结果判定:根据培养基上的微生物生长情况来判断样品是否受到微生物污染,需要操作者具备相关的判定能力。
7.记录和报告:对无菌检查结果进行准确地记录和报告,需要操作者具备良好的记录和沟通能力。

新版《中国药典》中关于无菌检查法的要点7月2日,国家药品监督管理局、国家卫生健康委发布公告,正式颁布2020年版《中国药典》,新版药典将于今年12月30日起正式实施。
新版药典对环境监测、无菌检查方法、微生物检测、灭菌验证等都做出了相关要求。
本文结合了《医疗器械无菌试验检查要点指南》中的检查内容,对新版《中国药典》中关于无菌检查法的要点进行了整理,供大家参考。
1、无菌检查要求(1)无菌检查应在无菌条件下进行,试验环境必须达到无菌检查的要求,检验全过程应严格遵守无菌操作,防止微生物污染,防止污染的措施不得影响供试品中微生物的检出。
(2)单向流空气区域、工作台面及受控环境应定期按医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法的现行国家标准进行洁净度确认。
隔离系统应定期按相关的要求进行验证,其内部环境的洁净度须符合无菌检查的要求。
(3)日常检验需对试验环境进行监测。
注意:“受控环境”主要的关注指标应该是悬浮粒子、浮游菌和沉降菌受控,其他如压差、温湿度等的指标和监测频度也应满足《药品洁净实验室微生物监测和控制指导原则》的要求。
2、培养基的制备及培养条件(1)培养基可使用按该处方生产的符合规定的脱水培养基或商品化的预制培养基。
配制后应采用验证合格的灭菌程序灭菌。
(2)制备好的培养基若不即时使用,应置于无菌密闭容器中,在2~25°C 、避光的环境下保存,并在经验证的保存期内使用。
注意:新版药典将“成品培养基”调整为“商品化的预制培养基”。
新版药典不再强调“非密闭-三周”、“密闭-一年”的规定,便于企业实践掌握;但企业应最好具备稳定的验证过程和结果,以支持封闭条件和保存周期。
3、培养基的适用性检查无菌性检查每批培养基一般随机取不少于 5 支(瓶),置各培养基规定的温度培养14 天,应无菌生长。
注意:应考虑实践中用于无菌性检查培养的试管装灭菌培养基与无菌检验用的三角瓶装培养基在灭菌方法验证时的两种包装的温度、F0等结果。

无菌检查法要点介绍及检验操作关键点培训1.无菌检查法是用来检验物体表面或物体内部是否有细菌或其他微生物存在的方法。
The aseptic inspection method is used to test whether there are bacteria or other microorganisms on the surface or inside of an object.2.无菌检查法要点包括选择合适的培养基、控制温度和湿度、以及严格的无菌操作。
The key points of aseptic inspection include selecting appropriate culture medium, controlling temperature and humidity, and strict aseptic operation.3.检验操作的关键点包括消毒操作、避免空气污染、以及避免人为污染。
The key points of inspection operation includedisinfection operation, avoiding air pollution, and avoiding artificial contamination.4.检查前需确保工作台面、器具和试剂都已进行消毒处理。
Before the inspection, it is necessary to ensure that the workbench, equipment, and reagents have been disinfected.5.操作人员要做好无菌操作前的准备工作,包括穿戴好无菌手套、口罩和工作服。
Operators should prepare for aseptic operation, including wearing sterile gloves, masks, and work clothes.6.在进行无菌操作时,要控制好空气流动,避免造成物体表面的污染。
1目的建立一个无菌检查法标准操作程序,保证实验的准确性、可靠性。
2范围适用于原料、辅料及成品的无菌检查。
3职责微生物检验员遵照执行。
4内容无菌检查法系用于检查药典要求无菌的药品、生物制品、医疗器具、原料、辅料及其他品种是否无菌的一种方法。
若供试品符合无菌检查法的规定,仅表明了供试品在该检验条件下未发现微生物污染。
无菌检查应在无菌条件下进行,试验环境必须达到无菌检查的要求,检验全过程应严格遵守无菌操作,防止微生物污染,防止污染的措施不得影响供试品中微生物的检出。
单向流空气区、工作台面及环境应定期按医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法的现行国家标准进行洁净度确认。
隔离系统应定期按相关的要求进行验证,其内部环境的洁净度须符合无菌检查的要求。
日常检验还需对试验环境进行监控。
4.1 培养基硫乙醇酸盐流体培养基主要用于厌氧菌的培养,也可用于需氧菌的培养;胰酪大豆胨液体培养基用于真菌和需氧菌的培养。
4.1.1 培养基的制备及培养条件培养基可按以下处方制备,亦可使用按该处方生产的符合规定的脱水培养基或成品培养基。
配制后应采用验证合格的灭菌程序灭菌。
制备好的培养基应保存在2〜25°C、避光的环境,若保存于非密闭容器中,一般在3 周内使用;若保存于密闭容器中,一般可在一年内使用。
4.1.1.1 硫乙酵酸盐流体培养基胰酪胨15. 0g 氯化钠 2. 5g酵母浸出粉 5. 0g 新配制的0. 1 % 刃天无水葡萄糖 5.0g 青溶液 1.0mlL-胱氨酸0. 5g 琼脂0. 75g硫乙醇酸钠0.5g 水1000ml(或硫乙醇酸)(0.3ml)除葡萄糖和刃天青溶液外,取上述成分混合,微温溶解,调节p H 为弱碱性,煮沸,滤清,加入葡萄糖和刃天青溶液,摇匀,调节p H , 使灭菌后在25℃的p H 值为7.1 土0.2。
分装至适宜的容器中,其装量与容器高度的比例应符合培养结束后培养基氧化层(粉红色)不超过培养基深度的1/2。