系统发育树构建
- 格式:ppt
- 大小:5.54 MB
- 文档页数:117

系统发育树的构建方法,使用的保守蛋白集
生物系统发育树(Phylogenetic tree)是分子生物学研究中最为常用的技术之一。
它可以预测到一组基因的演化过程,以便了解其衍生的生物类别的相对关系。
在构建生物系统发育树的过程中,常常使用保守蛋白集(conserved protein set)。
保守蛋白集是指在不同物种之间具有稳定序列并能够执行特定生物功能的蛋白质。
选择保守蛋白集作为建立生物系统发育树的分子标志物,这是因为它在沿着一个演化过程中保持稳定性,可以为树的构建提供有效的信息和数据。
此外,由于保守蛋白集通常都可以完全鉴定出来,而且序列之间的相似性要大于其它蛋白质,因此可以更加准确地定量表征这些物种的相似性。
在构建生物系统发育树时,首先要收集尽可能多物种的保守蛋白质序列,其次要对所有序列进行比较,然后用这些比较结果来构建一棵生物系统发育树。
其中,比较过程可以基于结构、功能、序列或者综合多种方法来完成,以便更准确地评估物种之间的相关性。
建立完成以后,可以提取从树中获得的信息来进一步研究这些物种的关系。
在生物系统发育树的构建过程中,使用保守蛋白集是一种有效的方法,它可以更准确地反映物种之间的关系,同时也有助于我们理解进化的模式和进程。
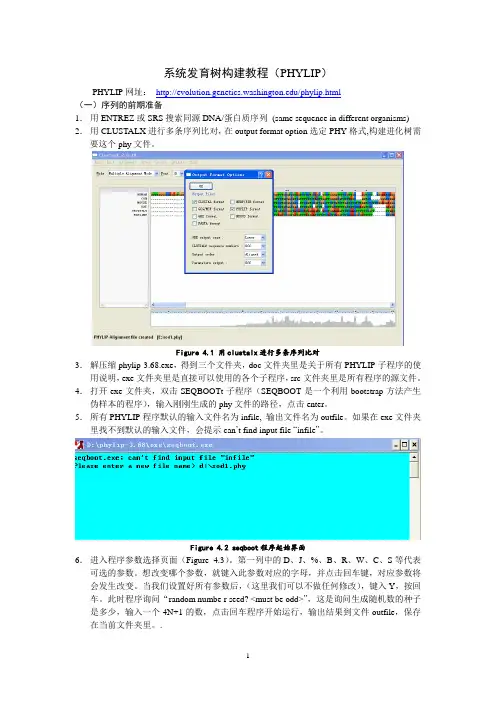
系统发育树构建教程(PHYLIP)PHYLIP网址:/phylip.html(一)序列的前期准备1.用ENTREZ或SRS搜索同源DNA/蛋白质序列(same sequence in different organisms) 2.用CLUSTALX进行多条序列比对,在output format option选定PHY格式,构建进化树需要这个phy文件。
Figure 4.1 用clustalx进行多条序列比对3.解压缩phylip-3.68.exe,得到三个文件夹,doc文件夹里是关于所有PHYLIP子程序的使用说明,exe文件夹里是直接可以使用的各个子程序,src文件夹里是所有程序的源文件。
4.打开exe文件夹,双击SEQBOOTt子程序(SEQBOOT是一个利用bootstrap方法产生伪样本的程序),输入刚刚生成的phy文件的路径,点击enter。
5.所有PHYLIP程序默认的输入文件名为infile, 输出文件名为outfile。
如果在exe文件夹里找不到默认的输入文件,会提示can’t find input file “infile”。
Figure 4.2 seqboot程序起始界面6.进入程序参数选择页面(Figure 4.3)。
第一列中的D、J、%、B、R、W、C、S等代表可选的参数。
想改变哪个参数,就键入此参数对应的字母,并点击回车键,对应参数将会发生改变。
当我们设置好所有参数后,(这里我们可以不做任何修改),键入Y,按回车。
此时程序询问“random numbe r seed? <must be odd>”,这是询问生成随机数的种子是多少,输入一个4N+1的数,点击回车程序开始运行,输出结果到文件outfile,保存在当前文件夹里。
.Figure 4.3 seqboot程序参数选择页面主要参数解释:D: 数据类型,有Molecular sequence、discrete morphology、restriction sites和gene frequencies4个选项。



分子系统发育树构建的简易方法
分子系统发育树的构建是根据分子序列的差异来推断不同物种之间的进化关系。
下面是一个简易的分子系统发育树构建方法:
1. 选择目标基因序列:选择与所研究物种相关的基因序列(如核糖体RNA或蛋白质编码基因)作为目标序列。
2. 数据收集:收集各个相关物种的目标基因序列数据。
可以通过公共数据库(如NCBI)或研究文献中的已有数据进行获取。
3. 序列比对:使用序列比对软件将收集到的序列进行比对,找出相同和不同的碱基或氨基酸位置。
常用的比对软件有CLUSTALW和MAFFT。
4. 构建进化树:根据序列比对结果,使用进化树构建软件(如MEGA)进行系统发育树的构建。
常用的进化树构建方法包括最大简约法(UPGMA)和最大似然法(ML)。
5. 进化树评估:对构建的系统发育树进行评估,可以使用Bootstrap方法进行支持值分析,提高树的可靠性。
6. 结果解读:根据构建的系统发育树,可以解读不同物种之间的进化关系和群体间的分化程度。
需要注意的是,分子系统发育树是基于目标基因序列的进化关系推断,仅仅代表目标基因的进化历史,并不一定能完全反映
整个物种的进化历史。
因此,在研究中还需要综合考虑其他重要因素,如形态特征和生态行为等。

多基因序列的系统发育树构建说到“多基因序列的系统发育树构建”这个话题,乍一听,可能有人会觉得这就是那种高深莫测、晦涩难懂的学术术语,甚至看一眼就头大。
其实嘛,说白了,这就像是在为大自然的大家族做一张族谱,揭开我们与其他物种之间千丝万缕的关系。
就像我们查家谱,看自己和曾祖父是不是同一个血统,看看自己和远方亲戚的亲疏。
要是能把这整个过程搞清楚了,哎,那可真是大开眼界,原来人类、植物、动物这些不同的生命形式之间,居然有那么多微妙又惊人的联系。
所谓的“系统发育树”就像是一本生命史诗,讲述的是各种物种之间的亲戚关系。
这棵树的根基上是我们共同的祖先,每一个分支代表了一条特定的进化路径。
而“多基因序列”呢,就是拿不同基因的信息去描绘这棵树的枝干,哪一枝长得快,哪一枝慢,这些都能通过基因序列的差异来看得一清二楚。
通俗点说,这就像是在给家谱里的每一位祖先添加更多的细节资料,越多的细节,越能精确地找到彼此之间的关系。
你看,这过程不就像拆谜题一样,一步步解开生物世界的神秘面纱吗?要构建这棵树,首先得有一堆基因数据。
别小看这些基因,它们可是真正的“家底”。
每个物种的DNA就像是一个个密码锁,里面藏着它们的生活历史、演化轨迹。
用这些信息,我们可以比较不同物种的基因,看看它们之间有多相似,或者差异有多大。
举个例子,人类和猴子的基因差异,真的是少得可怜,但这不代表我们是完全一样的。
那些微小的差异,往往就决定了我们是直立行走,还是蹦蹦跳跳。
所以呢,基因序列越多,越能描绘出一张更加真实、精准的系统发育树。
然后,咱们得选基因。
这不就是考古学家挑选遗骨进行复原的过程吗?我们得找那些能体现物种间差异的“好基因”。
这些基因应该既能反映物种的特性,又能体现进化的步伐。
选好了基因,接下来就要对它们做一番精细的比对。
这就好比你拿着一本古老的书,逐字逐句地对照,看这些字母和符号有没有相同或者不同。
这个过程需要非常细致,要小心翼翼,不容一丝疏忽。

系统发育树构建的三种方法
1. 距离法(Distance Method):该方法将各个物种之间的差异转化为距离值,并根据这些距离值构建系统发育树。
距离可以基于基因序列或形态特征等进行计算。
该方法不考虑进化模式和序列的进化过程,仅提供基于相似性的分支结构。
2. 最大简约法(Maximum Parsimony):该方法基于最小进化原则,即最可能的树是具有最少次数的进化事件的树。
它寻求在进化树上使得进化事件(如插入、缺失、突变)的次数最少的树。
该方法是需要较多计算的方法,但树的建立结果更加准确。
3. 最大似然法(Maximum Likelihood):该方法也是基于最小进化原则,但它考虑进化模式和序列的进化过程,并将最可能的进化树视为产生的序列数据的最大概率估计。
该方法需要更复杂的计算,但对于数据信息的准确推断较好。

叙述系统发育树的构建过程嘿,咱今儿就来讲讲系统发育树的构建过程,这可有意思啦!你看啊,系统发育树就像是一棵大树,它的枝桠代表着各种生物之间的关系。
那怎么把这棵大树给“种”出来呢?首先得有一堆生物的数据呀,就像盖房子得有砖头一样。
这些数据可以是各种各样的,比如基因序列啦、形态特征啦等等。
然后呢,就开始比对这些数据,这就好比把不同的砖头摆在一起,看看哪些相似,哪些不同。
接着,就根据这些比对的结果来确定它们之间的亲缘关系。
这就好像在给砖头们找它们的“家族”一样,哪些是近亲,哪些是远亲。
这可不是一件容易的事儿啊,得非常仔细地去分析。
然后呢,把这些亲缘关系用一种特别的方式表示出来,就像把砖头们按照一定的规律摆好,形成一个结构。
这个结构慢慢就变成了系统发育树的雏形。
这时候,就像是在给大树修剪枝叶一样,要对这个雏形进行调整和优化。
要确保每个部分都放对了位置,不能有差错。
最后,一棵完整的系统发育树就出来啦!哇塞,你想想看,通过这么多复杂的步骤,终于把生物之间的关系给清楚地呈现出来了,这难道不神奇吗?你说,这系统发育树构建的过程,像不像一个艺术家在精心雕琢一件作品?每一个细节都要处理好,才能呈现出完美的结果。
而且啊,这可不是一次性就能完成的事儿,得反复地去研究、去调整。
你再想想,要是没有系统发育树,我们怎么能知道各种生物之间有着这样那样的联系呢?我们怎么能更好地理解生命的奥秘呢?所以啊,这个构建过程虽然复杂,但真的超级重要呢!咱平时生活中也有类似的情况呀,比如说搭积木,不也是一块一块地搭起来,最后形成一个完整的造型嘛。
这和构建系统发育树不是有点像嘛!总之呢,系统发育树的构建过程就是这么神奇又有趣,它让我们对生物的世界有了更深的了解和认识。
这可真是一项伟大的工作啊!你难道不这么觉得吗?。
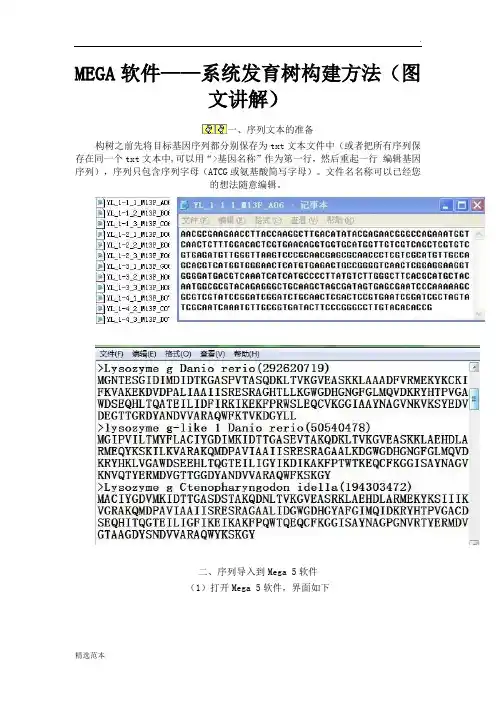
MEGA软件——系统发育树构建方法(图
文讲解)
一、序列文本的准备
构树之前先将目标基因序列都分别保存为txt文本文件中(或者把所有序列保存在同一个txt文本中,可以用“>基因名称”作为第一行,然后重起一行编辑基因序列),序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您
的想法随意编辑。
二、序列导入到Mega 5软件
(1)打开Mega 5软件,界面如下
(2)导入需要构建系统发育树的目的序列
OK
选择分析序列类型(如果是DNA序列,点击DNA,如果是蛋白序列,点击Prot
ein)
出现新的对话框,创建新的数据文件
选择序列类型
导入序列
导入序列成功。
(3)序列比对分析
点击工具栏中“W”工具,进行比对分析,比对结束后删除两端不能够完全对齐
碱基
(4)系统发育分析
关闭窗口,选择保存文件路径,自定义文件名称
三、系统发育树构建
根据不同分析目的,选择相应的分析算法,本例子以N—J算法为例
Bootstrap 选择1000,点击Compute,开始计算
计算完毕后,生成系统发育树。
.
根据不同目的,导出分析结果,进行简单的修饰,保存
精选范本。

构建系统发育树的方法
构建系统发育树的方法
一、定义
系统发育树(Phylogenetic Tree)又称为系统种群学树,是一
种描述物种演化的树型结构,从根节点开始描述物种主要进化分支结构,树上的每条边则表示两个物种在进化的历史中距离彼此更近或来自同一进化祖先的关系。
二、建立系统发育树的方法
1.收集数据:系统发育树的建立首先要收集数据,作为建立树的基础,这些数据一般是利用各种实验技术来收集,比如遗传学实验和物种形态的实验。
2.选取特征:从收集的大量数据中,应选取尽可能多的可靠特征,作为建立树的材料,这些特征要有规律性,有可靠性,可以容易发现物种之间的内在关系,有利于在研究中可靠地比较各物种之间的相似程度。
3.分类比较:将所有待比较的物种或实体按照类似的特征进行分类,根据同一物种种的特征之间的差异,可以比较出物种之间的相似度,确定出有利于建立树的特征。
4.描绘树枝:根据比较的结果,可以依次将物种分类编码,从根节点开始,逐级分细枝条,最后得出系统发育树的图形结构。
5.校正树枝:检查系统发育树的构建结果,如果发现有一些物种不太符合物种演化过程的规律,可以根据其他数据和结果来校正树枝,
从而得出最终的发育树结构。
构建系统发育树需要注意的几个问题1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。
2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。
3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。
4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。
5 枝长可以用来表示类间的真实进化距离。
6 重要的是理解系统发育分析中的计算能力的限制。
任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。
7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。
然而,有些方法可以检测系统发育树检测的可靠性。
第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。
分子进化研究的基本方法对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。
表型的(phenetic)和遗传的(cladistic)数据有着明显差异。
Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。
这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。
表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。
进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。
文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。
系统进化树分有根(rooted)和无根(unrooted)树。
系统发育树构建的三种方法
系统发育树(Systems 发育 Tree,简称Stree)是一种用于描述生物系统进化的图形化工具,通常用于模拟生物系统行为的演化过程。
以下是三种构建系统发育树的方法:
1. 基于规则的方法:这种方法使用预定义的规则和偏好来构建
系统发育树。
例如,可以使用遗传算法或人工神经网络等机器学习方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法需要大量
的人工工作,但可以生成较为准确的演化树。
2. 基于统计方法的方法:这种方法使用统计学方法来推断物种
之间的演化关系。
例如,可以使用最大似然估计或贝叶斯推断等方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法不需要人工工作,但需要更多的计算资源和时间,才能得到比较准确的演化树。
3. 基于模型的方法:这种方法使用已经建立的模型和数据来构
建系统发育树。
例如,可以使用层次结构模型(如生物进化树、社会网络模型等)来预测一个物种的遗传特征或行为演化轨迹。
这种方法可
以快速构建系统发育树,但需要更多的人工工作来验证模型的准确性。
系统发育树的数字一、什么是系统发育树系统发育树(Phylogenetic tree)是生物学中常用的一种图形表示方式,用于展示不同物种之间的进化关系。
通过构建系统发育树,我们可以了解物种之间的亲缘关系、进化历史以及共同祖先等重要信息。
系统发育树的构建是基于物种间的共有衍征特征以及遗传信息进行的。
二、系统发育树的构建方法2.1 形态学特征比较法形态学特征比较法是通过对不同物种的形态特征进行比较,从而推断它们之间的亲缘关系。
这种方法适用于无法获取遗传信息的化石物种或者某些现存物种。
通过比较形态特征的相似性和差异性,可以推断物种之间的进化关系。
2.2 分子生物学方法分子生物学方法是目前构建系统发育树最常用的方法之一。
这种方法利用DNA、RNA或蛋白质序列的比较,推断物种之间的亲缘关系。
通过比较序列的相似性和差异性,可以构建出更加准确的系统发育树。
2.3 综合方法综合方法是将形态学特征比较法和分子生物学方法相结合,以获取更全面和准确的系统发育信息。
这种方法可以同时考虑形态特征和遗传信息,从而得出更可靠的系统发育树。
三、系统发育树的数字表示系统发育树的数字表示是为了更直观地展示物种之间的亲缘关系和进化距离。
在系统发育树中,每个物种都被表示为一个节点,节点之间的连接线表示它们之间的进化关系。
系统发育树的数字表示主要包括以下几个方面:3.1 分支长度分支长度表示物种之间的进化距离。
通常情况下,分支长度越长,表示物种之间的进化距离越远;分支长度越短,表示物种之间的进化距离越近。
通过分支长度的比较,我们可以了解不同物种之间的进化速度和差异程度。
3.2 节点标签节点标签表示每个节点所代表的物种名称。
通过节点标签,我们可以清楚地了解每个物种在系统发育树中的位置和亲缘关系。
3.3 分支支持率分支支持率表示对系统发育树分支的支持程度。
分支支持率越高,表示该分支的构建更加可靠和准确;分支支持率越低,表示该分支的可靠性较低。
三种方法构建系统发育树学习笔记所用数据为一个属内不同种不同群体的叶绿体基因组序列,数量为80条。
发现用全长序列建树的时候,不适合选用太多外类群,否则ML法中会导致属内分枝的枝长特别短。
原因应该是基因间隔区和内含子区域序列位点的差异较大。
枝长含义NJ:表示遗传距离;MP:性状状态变换的替换数;ML/BI:该分枝上的相对进化数量(遗传变异量);每个位点上的替换数(一般以每位点多少次核苷酸替换或氨基酸取代来表示)。
遗传距离大多数情况以序列来说遗传距离就是两个OTU(个体、群体、物种或基因家族)之间序列的差异值。
序列比对多序列比对用mafft得到的结果较为准确,muscle比对的速度较快。
多序列比对的绝大多数算法都是基于渐进比对的概念。
简单来说就是先从两个序列的比对开始,逐渐添加新序列,直到所有的序列都加入为止。
但是不同的添加顺序会产生不同的比对结果。
所以由最相似的两个序列开始比对,由近到远逐步完成最为可靠。
mafft --thread 15 --auto 80-AcoeOut.fasta > 80-AcoeOut_aln.fasta##比对时如果不清楚什么参数合适,加个参数--auto,软件可以自动帮你处理挑选保守位点进行下一步建树序列比对完后,用于建树的序列位点必须保证具有良好的同源性。
所以需要删除序列分歧很大的区域和gap区域。
我用的软件为Gblocks,主要目的是把有gap的位点全部去除,参数为-b5=n,其余的选项有-b5=h,h表示half 指去除在大于50%的序列中出现gap的位点。
Gblocks 80-AcoeOut_aln.fasta -t=d -b5=n最大简约法(软件PAUP)最大简约法的树长指所有性状在一棵树上的进化改变总数。
计算得到的结果可能会有许多树长相等的简约树,此时需要计算它们的一致树。
分为strict consensus和semistrict consensus等,strict表示100%,在所有简约树中都出现的分枝,才会出现在一致树中,否则为梳子。
系统发育树的构建方法
嘿,朋友们!今天咱来聊聊系统发育树的构建方法,这可有意思啦!
你想想啊,系统发育树就像是一棵大树,上面挂满了各种生物,它们之间有着千丝万缕的联系。
那怎么把这棵大树给建起来呢?
首先得有数据呀!就像盖房子得有砖头一样。
这些数据可以是各种生物的特征啦、基因序列啦等等。
这可不能马虎,得仔细收集。
然后呢,就是选择合适的方法啦。
这就好比做菜,得选对调料和烹饪方法才能做出美味的菜肴。
不同的方法有不同的特点,得根据实际情况来选。
接下来,就是分析数据啦!这就像是侦探在破案,要从一堆线索中找出真相。
得仔细琢磨每个数据的意义和关系。
在这个过程中,可不能瞎搞哦!得有耐心,就像绣花一样,一针一线都要精细。
要是马马虎虎,那建出来的树可就歪七扭八啦。
还有哦,要不断地调整和优化。
就像雕刻一件艺术品,得不断地打磨才能让它更完美。
建系统发育树可不是一件容易的事儿,但当你看到那棵清晰地展示出生物之间关系的大树时,那种成就感,哇,简直没法形容!
你说,这是不是很神奇?通过这样的方法,我们就能更好地了解生物的演化历程,就像穿越时空看到了它们的过去一样。
这不就像是我们在探索一个神秘的世界吗?每一个数据都是一个线索,每一次分析都是一次冒险。
所以啊,朋友们,别小看了系统发育树的构建方法,它可是打开生物奥秘大门的一把钥匙呢!让我们一起努力,去构建出更漂亮、更准确的系统发育树吧!
原创不易,请尊重原创,谢谢!。