最新手把手教你构建系统进化树
- 格式:ppt
- 大小:2.79 MB
- 文档页数:7

构建进化树的步骤通常包括以下几个关键环节:
1. 数据收集:收集相关的生物序列数据,这些数据可以来自于公共数据库,如NCBI的GenBank,也可以通过实验获得。
序列数据包括DNA或蛋白质序列。
2. 序列alignment(序列比对):使用比对软件如Clustal Omega、MAFFT、MUSCLE等,将收集到的序列进行比对,以确保序列的同源性,并消除由于序列变异导致的噪音。
3. 序列拼接和校正:对测序得到的正向和反向序列进行拼接和校正,以获得完整的序列。
常用的拼接软件有Contig Express、Geneious 和Sequencher等。
4. 选择合适的模型:根据序列数据选择合适的进化模型。
可以使用软件如Modeltest来评估不同的进化模型,选择BIC(Bayesian Information Criterion)分数最低的模型。
5. 建树:选择合适的软件和建树方法来构建进化树。
常用的软件有MEGA、PhyML、MrBayes等,建树方法包括NJ(邻接法)、MP (最大简约法)、ML(最大似然法)等。
6. 建树检验:使用如Bootstrap方法等来检验所建树的稳定性和可靠性。
Bootstrap方法通过重复抽样来检验建树的节点支持度。
7. 绘制进化树:使用软件如TreeDraw、FigTree或在线工具来绘制进化树的图像,以便于分析和展示。
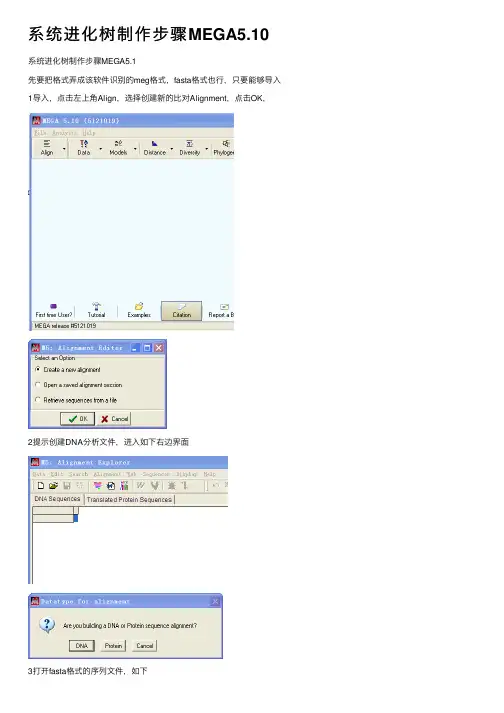
系统进化树制作步骤MEGA5.10系统进化树制作步骤MEGA5.1
先要把格式弄成该软件识别的meg格式,fasta格式也⾏,只要能够导⼊1导⼊,点击左上⾓Align,选择创建新的⽐对Alignment,点击OK,
2提⽰创建DNA分析⽂件,进⼊如下右边界⾯
3打开fasta格式的序列⽂件,如下
4选择Alignment中的⽐对选项,使⽤Clastaw⽐对,会有⽐对参数,默认即可,点OK,会⾃⾏完成⽐对。
5⽐对好的⽂件须保存,选Data中的save session,提⽰保存命名
即导⼊数据如右。
7选中带有TA的⽅框,如上图,点击主界⾯上的进化树选项,如下图中间,可选择不同的进化树类型,⼀般⽤NJ树。
8出现提⽰参数,默认即可,点compute执⾏。
会完成进化树
9做好的进化树可以以图⽚⽅式保存。

系统进化树的建立(完整)1. 进化树的建立软件:MEGA输入文件:fas格式文件输出文件:nwk格式文件建立过程1)将要用于构建系统进化树的所有序列合并到同一个fasta格式文件;2)打开MEGA软件,选择主窗口的”File” → “Open A File”→找到并打开fasta文件,这时会询问以何种方式打开,需要先进行多序列比对,所以选择“Align”。
如果是比对好的多序列比对可以直接选择“Analyze”。
3)打开的Alignment Explorer窗口中选择”Alignment”“Align by -ClustalW” 进行多序列比对,弹出窗口询问“Nothing selected for alignment,Select all?”选择“OK”。
4)之后,弹出多序列比对参数设置窗口。
这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。
MEGA的所有默认参数都是经过反复考量设置的,这保证了MEGA傻瓜机全自动档的品质,所以当你无从下手,或者没有什么特别要求的时候,直接点击“OK”,接受这些默认参数,开始多序列比对。
5)比对过程是先进行双序列比对,在进行多序列比对,最后会出现一个多序列比对结果。
将之作为中间结果保存下来。
在Alignment Explorer窗口中选择“Data”→“Export Alignment”→选择要保存文件的格式(一般用meg格式)4)生成的“.meg”文件可以双击直接导入MEGA。
点击data-Phylogenetic Analysis,回到MEGA主界面。
5)开始建树。
点击MEGA主窗口上的Phylogeny下拉菜单,选择Neighbor Joining(最近邻居法)。
保存为nwk格式文件2.进化树美化软件:Rstudio(ggtree包)输入文件:nwk文件输出文件:建立的彩色进化树美化过程R语言代码:#加载R包install.packages('ggtree')install.packages('ggplot2')library(ggplot2)library(ggtree)#读取树文件x <- read.tree('***自己的文件(一定注意路径***)')#读取分组信息groupInfo <- split(x$bel, gsub('_\\w+', '', x$bel))#按类分组y <- groupOTU(x, groupInfo)#将分组信息添加到树中tree <- groupOTU(x, groupInfo)#绘制进化树ggtree(tree, layout='fan', ladderize = FALSE, branch.length = 'none',aes(color=group)) + geom_tiplab2(size=3) + theme(legend.position = 'right')。

构建系统进化树的方法步骤1. 建树前的准备工作1.1 相似序列的获得——BLASTBLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。
国际著名生物信息中心都提供基于Web的BLAST服务器。
BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为内核向两端延伸,以找出尽可能长的相似序列片段。
首先登录到提供BLAST服务的常用网站,比如国内的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。
这些网站提供的BLAST服务在界面上差不多,但所用的程序有所差异。
它们都有一个大的文本框,用于粘贴需要搜索的序列。
把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。
如果是DNA序列,一般选择BLASTN搜索DNA数据库。
这里以NCBI为例。
登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。
BLASTN结果如何分析(参数意义):>gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, complete sequenceScore = 2020 bits (1019), Expect = 0.0Identities = 1382/1497 (92%), Gaps = 8/1497 (0%)Strand = Plus / PlusQuery: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60 |||||||||||||||||||||||||||||||||||||||||| ||||||||| |||||Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120 || ||||||||||||||||||||||||||||||| | |||||| |||||||||||||Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118Score :指的是提交的序列和搜索出的序列之间的分值,越高说明越相似;Expect:比对的期望值。


大家好:我在此介绍几个进化树分析及其相关软件的使用和应用范围。
这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN(LINUX)。
在介绍软件之前,我先简要地叙述一下有关进化树分析的一些方法学问题。
进化树也称种系树,英文名叫“Phyligenetic tree”。
对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行排列(To align sequences)。
做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。
⑵要构建一个进化树(To reconstrut phyligenetic tree)。
构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。
所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。
而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。
进化树枝条的长度代表着进化距离。
独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。
⑶对进化树进行评估。
主要采用Bootstraping法。
进化树的构建是一个统计学问题。
我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。
如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。

大家好:我在此介绍几个进化树分析及其相关软件的使用和应用范围。
这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN (LINUX)。
在介绍软件之前,我先简要地叙述一下有关进化树分析的一些方法学问题。
进化树也称种系树,英文名叫“Phyligenetic tree”。
对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行排列(To align sequences)。
做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。
⑵要构建一个进化树(To reconstrut phyligenetic tree)。
构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。
所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。
而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。
进化树枝条的长度代表着进化距离。
独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。
⑶对进化树进行评估。
主要采用Bootstraping法。
进化树的构建是一个统计学问题。
我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。
如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。

构建系统发育树的三大方法
1、距离法:基于距离的方法,首先通过各个物种之间的比较,根
据一定的假设(进化距离模型)推导得出分类群之间的进化距离,构建一个进化距离矩阵。
进化树的构建则是基于这个矩阵中的进化距离关系。
2、特征法:基于特征的方法,不计算序列间的距离,而是将序列
中有差异的位点作为单独的特征,并根据这些特征来建树。
3、简约法:基于简约的方法,通过构建一棵由所有可能的子树组
成的树,然后从这个树中选择一个最优的子树作为进化树。

大家好:我在此介绍几个进化树分析及其相关软件的使用和应用范围。
这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN(LINUX)。
在介绍软件之前,我先简要地叙述一下有关进化树分析的一些方法学问题。
进化树也称种系树,英文名叫“Phyligenetic tree”。
对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行排列(To align sequences)。
做ALIGNMENT的软件很多,最经常使用的有CLUSTALX 和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。
⑵要构建一个进化树(To reconstrut phyligenetic tree)。
构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。
所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。
而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。
进化树枝条的长度代表着进化距离。
独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。
⑶对进化树进行评估。
主要采用Bootstraping法。
进化树的构建是一个统计学问题。
我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。
如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。

构建系统进化树的详细步骤1. 建树前的准备工作1.1 相似序列的获得——BLASTBLAST是目前常用的数据库搜索程序,它是Basic Local Alignment Search Tool 的缩写,意为“基本局部相似性比对搜索工具”(Altschul et al.,1990[62];1997[63])。
国际著名生物信息中心都提供基于Web的BLAST服务器。
BLAST算法的基本思路是首先找出检测序列和目标序列之间相似性程度最高的片段,并作为核向两端延伸,以找出尽可能长的相似序列片段。
首先登录到提供BLAST服务的常用,比如国的CBI、美国的NCBI、欧洲的EBI和日本的DDBJ。
这些提供的BLAST服务在界面上差不多,但所用的程序有所差异。
它们都有一个大的文本框,用于粘贴需要搜索的序列。
把序列以FASTA格式(即第一行为说明行,以“>”符号开始,后面是序列的名称、说明等,其中“>”是必需的,名称及说明等可以是任意形式,换行之后是序列)粘贴到那个大的文本框,选择合适的BLAST程序和数据库,就可以开始搜索了。
如果是DNA序列,一般选择BLASTN搜索DNA数据库。
这里以NCBI为例。
登录NCBI主页-点击BLAST-点击Nucleotide-nucleotide BLAST (blastn)-在Search文本框中粘贴检测序列-点击BLAST!-点击Format-得到result of BLAST。
BLASTN结果如何分析(参数意义):>gi|28171832|gb|AY155203.1| Nocardia sp. ATCC 49872 16S ribosomal RNA gene, completesequenceScore = 2020 bits (1019), Expect = 0.0Identities = 1382/1497 (92%), Gaps = 8/1497 (0%) Strand = Plus / PlusQuery: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggaaaggccctttcgggggt 60|||||||||||||||||||||||||||||||||||||||||| ||||||||| ||||| Sbjct: 1 gacgaacgctggcggcgtgcttaacacatgcaagtcgagcggtaaggcccttc--ggggt 58Query: 61 actcgagcggcgaacgggtgagtaacacgtgggtaacctgccttcagctctgggataagc 120|| ||||||||||||||||||||||||||||||| | |||||| ||||||||||||| Sbjct: 59 acacgagcggcgaacgggtgagtaacacgtgggtgatctgcctcgtactctgggataagc 118Score :指的是提交的序列和搜索出的序列之间的分值,越高说明越相似; Expect:比对的期望值。

系统进化树构建方法及软件应用系统进化树是用来描述生物物种间亲缘关系的图表化工具,可以通过比较不同物种的遗传信息来确定它们之间的关系。
构建系统进化树可以帮助研究人员理解生物多样性的起源和发展。
本文将介绍系统进化树的构建方法,并介绍一些常用的软件应用。
构建系统进化树的方法主要分为两大类:演化模型和系统发育理论。
演化模型是基于遗传信息的演化过程进行建模,并通过统计学方法比较不同物种之间的遗传差异。
系统发育理论则是根据具体的分类原则和假设来分析和解释不同物种之间的关系。
下面将详细介绍一些常用的构建系统进化树的方法:1.分子钟模型:分子钟模型是一种基于遗传物质的演化模型,通过比较物种间的遗传差异,并根据时间尺度来估计各物种分化的时间。
分子钟模型主要依赖于分子演化速率的恒定性假设,即物种间的多态性和突变速率是恒定的。
这种方法广泛应用于研究不同物种的分子进化关系。
2.最大似然法:最大似然法是一种常用的计算统计学方法,通过计算在给定模型条件下观测到的数据(例如DNA序列)的概率来估计系统进化树。
该方法假设不同物种的进化关系可以用一个概率模型来表示,并通过调整模型参数来最大化观测序列出现的概率。
3.距离法:距离法是一种直接测量不同物种间的遗传距离(即序列差异)的方法。
它基于分子进化或形态特征的测量来生成系统进化树。
距离法没有明确的进化模型,常用的计算方式包括简约性方法和邻居法。
除了上述的构建系统进化树的方法,还有一些软件应用可以帮助研究人员进行系统进化树的构建和分析。
下面介绍几个常用的软件应用:1.MEGA:MEGA是一款广泛使用的分子进化分析软件,提供了多种方法来构建系统进化树,包括最大似然法、贝叶斯方法和邻居法等。
它还提供了一系列的工具来分析进化树的可靠性和比较不同分支的进化速率。
2.PAUP*:PAUP*是一款用于构建系统进化树的软件,它提供了多种分析方法和模型选择工具,可以根据研究需要选择适当的方法和模型。
系统进化树的构建1. 引言在计算机科学领域,系统进化树是一种用于描述和分析软件系统演化历史的工具。
它可以帮助我们理解软件系统是如何随着时间发展和演变的,以及不同版本之间的关系。
通过构建系统进化树,我们可以更好地了解软件系统的演化规律,为软件维护、升级和迭代提供有效的指导。
本文将详细介绍系统进化树的构建方法,并提供相关示例和实践经验。
2. 构建方法2.1 数据收集构建系统进化树的第一步是收集相关数据。
这些数据可以来自于版本控制系统、缺陷跟踪系统、代码仓库等多个来源。
主要包括以下几个方面:•版本信息:记录每个版本的发布日期、版本号等基本信息。
•变更集:记录每个版本中进行了哪些变更,包括新增功能、修改bug等。
•缺陷报告:记录每个版本中出现的缺陷报告,包括缺陷编号、严重程度等。
•代码仓库:记录每个版本中所使用的代码库快照。
2.2 数据预处理在进行数据分析之前,需要对收集到的数据进行预处理。
主要包括以下几个方面:•数据清洗:去除重复、无效或不完整的数据。
•数据整合:将不同来源的数据进行整合,建立关联关系。
•数据格式化:将数据转换为统一的格式,方便后续分析和处理。
2.3 构建演化关系构建系统进化树的核心是建立不同版本之间的演化关系。
可以使用以下两种方法来实现:2.3.1 基于变更集通过分析每个版本中的变更集,可以识别出新增、修改和删除的功能模块或代码文件。
根据这些变更信息,可以构建出一个版本间的差异图,从而揭示出系统演化的路径。
2.3.2 基于缺陷报告通过分析每个版本中出现的缺陷报告,可以识别出哪些缺陷被修复,并确定修复缺陷所涉及到的代码文件或功能模块。
根据这些信息,可以构建出一个修复路径图,从而揭示系统演化过程中缺陷修复的路径。
2.4 可视化展示构建完成系统进化树后,需要将其以可视化形式展示出来。
常用的可视化工具有网络图、树状图等。
通过可视化展示,可以更直观地了解系统的演化历史和各个版本之间的关系。
3. 示例与实践经验3.1 示例以一个开源软件项目为例,假设我们收集到了该项目的版本控制记录、缺陷报告和代码仓库快照。
大家好:我在此介绍几个进化树分析及其相关软件的使用和应用范围。
这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN (LINUX)。
在介绍软件之前,我先简要地叙述一下有关进化树分析的一些方法学问题。
进化树也称种系树,英文名叫“Phyligenetic tree”。
对于一个完整的进化树分析需要以下几个步骤:⑴要对所分析的多序列目标进行排列(To align sequences)。
做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。
⑵要构建一个进化树(To reconstrut phyligenetic tree)。
构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。
所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。
而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。
进化树枝条的长度代表着进化距离。
独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。
⑶对进化树进行评估。
主要采用Bootstraping法。
进化树的构建是一个统计学问题。
我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。
如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。
系统进化树的构建方法
利用最大似然法构建水稻蛋白系统进化树
1 蛋白序列获取
在MSU上检索蛋白序列,最终检索获取到770条蛋白序列。
2 多序列比对
利用CLUSTALW 2.1软件(Higgins DG等,2007),使用默认参数,对770条蛋白序列进行多序列比对。
3 构建系统进化树
利用IQ-TREE 1.6.12构建系统进化树。
使用最大似然法(Maximum Likelihood,ML),设置自动测试546种替换模型,最终选用最优替换模型JTT+R10,bootstrap重复1000次。
4 绘制系统进化树
利用iTOL 5.4(Ivica Letunic等,2006),绘制系统进化树,删除bootstrap value低于70的分枝。
5 检验
参考Guo-Liang Wang(2011)、Hong-Wei Xue(2012)、Narendra Tuteja(2012)、Ki-Hong Jung(2014)等发表的关于某蛋白的文章,将770条蛋白系统进化树与文献中的系统进化树进行比较,结果基本一致。